DOI:
10.1039/C1RA00461A
(Paper)
RSC Adv., 2011,
1, 1050-1059
Synthesis of lactones of ortho-tyrosine, DOPA isomers and tryptophan-ortho-tyrosine hybrid amino acids†
Received
15th July 2011
, Accepted 19th July 2011
First published on 8th September 2011
Abstract
A new high-yielding syntheses of lactones of ortho-tyrosine, four isomers of DOPA and a hybrid amino acid incorporating tryptophan and ortho-tyrosine have been achieved in a two step sequence from 2-methylamino-3-nitro-4-methylsulfanyl 4H-chromenes 7. Substitution of the C4 methylsulfanyl group in 7 with hydrogen or with electron rich aromatic compounds furnished corresponding 2-methylamino-3-nitro-4H-chromenes. Reduction of the nitro group with concomitant hydrolysis of the enamine moiety in 2-alkylamino-3-nitro-4H-chromenes or in 4-aryl-2-alkylamino-3-nitro-4H-chromenes with Zn, Ac2O and AcOH led to facile synthesis of lactones of ortho-tyrosine, four isomers of DOPA and the hybrid amino acid incorporating tryptophan and ortho-tyrosine units.
Introduction
Non-essential and hybrid amino acids hold much promise as therapeutic agents.1 For example, ortho-tyrosine 1 (Fig. 1), a non-essential amino acid and synthesized in the body from phenylalanine, has been proposed as a marker for identification of the patients affected with Diabetes mellitus.2 Moreover, patients affected with chronic kidney diseases produce ortho-tyrosine in larger quantities, levels of which in urine can be used for detection of these diseases.3ortho-Tyrosine and its lactone 2 (Fig. 1) are formed in the body through non-enzymatic free radical hydroxylation of phenylalanine under conditions of oxidative stress and at the time of radiolytic oxidation of proteins.4 During limited radiolysis, their concentration in proteins increases gradually as a function of absorbed dose.5 So, ortho-tyrosine has been proposed as a marker for the identification of irradiated protein-rich food.6
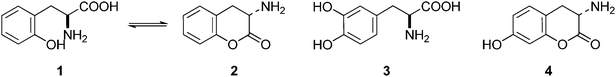 |
| | Fig. 1 Structures of ortho-tyrosine 1, ortho-tyrosine lactone 2, dihydroxyphenylalanine (DOPA) 3 and the lactone derived from an isomer of DOPA 4. | |
Dihydroxyphenylalanine (DOPA, Fig. 1) 3 is an essential amino acid. It has a major role in neuro-transmittance and is used for the treatment of Parkinson’s disease.7,8 However, the role of isomers of DOPA, which are non-natural amino acids or their lactones (Fig. 1) is little understood as the availability of these compounds is scarce. Thus, large scale and convenient synthesis of ortho-tyrosine and the isomers of DOPA or their lactonese.g.4 (Fig. 1) will facilitate to comprehend their biological role. Hybrid amino acids are valuable tools for understanding of the roles and functions of the natural amino acids.9 Incorporation of hybrid amino acids into predesigned proteins would greatly expand the ability to rationally and systematically manipulate their structure and probe their function. By this process, it is possible to modify existing proteins or create artificial proteins with new properties.10
We have recently shown that 2-methylamino-3-nitro-4H-chromenes 7 can be synthesized readily and rapidly in quantitative yield from 2-hydroxybenzaldehydes 5 and N-alkyl-N-[1-(methylsulfanyl)-2-nitro-1-ethenyl]amines (for e.g. 1N-methyl-1S-methyl-2-nitroethylene; NMSM) 6 through atom economic condensation reaction (Scheme 1).11 The C4-SMe group in 4H-chromenes 7 was found to be labile and it could be replaced with aryl/alkyl thiols like thiophenol or octane thiol. We reasoned that reductive removal of C4-SMe group in 7 should provide 4H-chromenes which on further reduction of the nitro group to amino followed by hydrolysis of the enamine functionality should provide lactones of amino acids like ortho-tyrosine and DOPA isomers. Similarly, replacement of SMe in 7 with electron rich aromatic compounds followed by reduction of the nitro group and hydrolysis of the enamino group should provide lactones of hybrid amino acids. Presently we report realization of this concept for a facile synthesis of lactones of ortho-tyrosine, four isomers of DOPA and a hybrid amino acid incorporating tryptophan and ortho-tyrosine units.
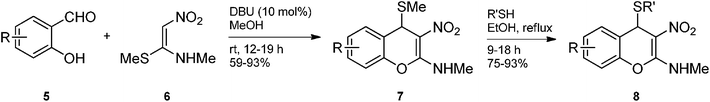 |
| | Scheme 1 Synthesis of 2-methylamino-3-nitro-4-alkylsulfanyl 4H-chromenes 7 and 8 | |
Results and discussion
Reductive removal of SMe in 4H-chromenes 7
Seemingly facile reductive removal of C4-SMe from 7a with Raney ® nickel proved to be capricious. Not only did the reaction depend on the batch of the Raney ® nickel employed, it also did not yield desired product N-methyl-3-nitro-4H-chromen-2-amine 12a (see Scheme 4). The reduction was attempted in different solvents like EtOH, EtOH-THF, THF or i-PrOH-THFetc., at −78 °C/0–5 °C/rt/reflux temperature of the solvent. In each case, either there was no reaction at low temperature or there was extensive decomposition of the reactants when conducted at reflux temperature of the solvent. Only in one run, we isolated a dimeric product 9 in 11% yield (Scheme 2). In one another run, the reaction provided N2-methyl-2H-2,3-chromenediamine 10 in 12% yield.
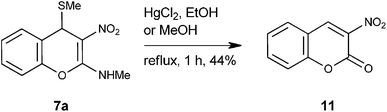
Reductive removal of SMe in 4H-chromene 7a did not take place with H2 and 10% Pd-C in presence or absence of acetic acid. Similarly hydride reducing agents like NaBH4 in MeOH, NaBH4 in EtOH, NaBH4 and BF3.Et2O in THF (BH3), LiAlH4 in dry diethyl ether at −78 °C or at rt did not yield any tangible product. Attempted desulfurization with HgCl2 in EtOH12 or catalytic amount of HCl in dry MeOH provided known 3-nitro-coumarin 1113 in moderate yield (Scheme 3).
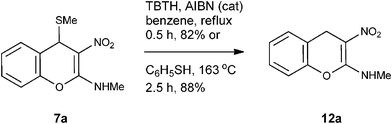
Failure of reductive desulfurization with H2/Pd-C or with hydride donors (both nucleophilic e.g.NaBH4 and electrophilic e.g.BH3, reducing agents) forced us to rethink on the alternative ways. We reasoned that if the SMe has to be removed reductively under acidic or basic conditions, concomitant decarboxylation of the exposed carboxylic acid was a clear possibility. Therefore we concluded that the SMe removal must be carried under radical or neutral conditions. As a first choice (Method A in the experimental section) we employed, tri-n-butyltin hydride (TBTH) in the presence of 2,2’-azobisisobutyronitrile (AIBN).14 This reaction proved to be inconsistent; when it worked, a maximum of 82% yield of 12a could be generated. After significant reaction optimization, we were elated to find that SMe removal did take place when 4H-chromene 7a was heated with thiophenol in a sealed tube at 163 °C in a pre-heated oil bath. If the reaction was conducted in open vessel; at least five equivalents thiophenol was required for complete conversion to 12a (Scheme 4).
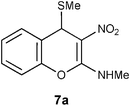
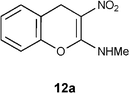
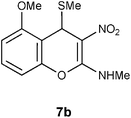
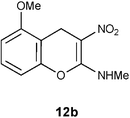
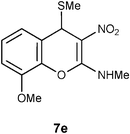
Mechanistically the above transformation could go through the formation of methyl phenyl disulfide to generate a carbanion intermediate, which gets quenched by the protic solvent. Employment of thiophenol for the above transformation required stringent condition of heating reaction mixture in obnoxiously smelling thiophenol (Method B in the experimental section). As an option, the biomimetic hydride donor, the Hantzsch diester appeared to be a suitable reagent for this transformation.15 Hantzsch diester, prepared over a century ago, mimics biological hydride donors like NADH and NADPH. It is acquiring increased attention in recent years as a selective and mild reducing agent operating at neutral conditions.16 We were happy to find that Hantzsch diester in MeOH reflux, converted 7a to 12a in 78% yield (Method C in the experimental section, Scheme 5). Reductive removal of SMe from 4H-chromene 7a by this method was proved to be general. We conducted this transformation on five different substrates and in each case 4H-chromenes 12b–e were obtained in good yield (Table 1).
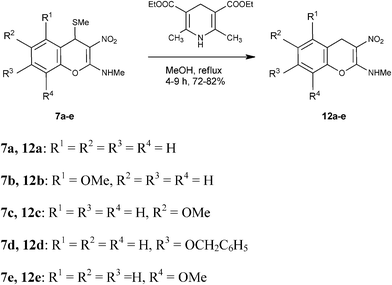 |
| | Scheme 5 Synthesis of 2-methylamino-3-nitro-4H-chromenes 12a–e from 2-methylamino-3-nitro-4-methylthio-4H-chromenes 7a–e using Hantzsch diester. | |
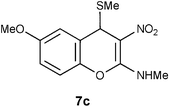
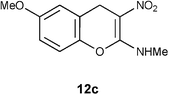
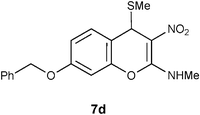
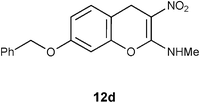
Table 1 Reductive removal of SMe in 2-methylamino-3-nitro-4-methylthio-4H-chromenes 7a–e using Hantzsch diester to form 4H-chromenes a–e.
| Entry |
Substrate |
Product |
Time (h) |
Yield (%) |
| 1 |
|
|
4 |
78 |
| 2 |
|
|
7 |
74 |
| 3 |
|
|
9 |
86 |
| 4 |
|
|
7 |
82 |
| 5 |
|
|
8 |
72 |
At this point it is worth noting an experiment conducted on the benzyl ether 7d with thiophenol reflux. Interestingly, unlike the reaction with other isomers like 7b–c or 7d this reaction did not yield any characterizable product. Similarly, corresponding allyl ether also did not undergo reductive desulfurization, indicating labile nature of the protecting group placed on the C7 carbon of 7. Reductive removal of SMe, from the benzyl ether 7d, however, worked well with Hantzsch diester (entry 4, Table 1).
Synthesis of lactones of ortho-tyrosine, DOPA isomers
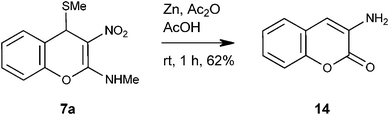
In view of the importance of ortho-tyrosine, we embarked to convert 4H-chromene 7a into ortho-tyrosine lactone 13a (Fig. 2).17 For this purpose it was required to reduce the nitro group in 12a to the amino group and hydrolyze the enamine functionality to the lactone carbonyl.18 Initially, we tried a single-step conversion of 7a to 13a by employing Zn, Ac2O and AcOH19 or Fe and AcOH.20 Both the reactions, however, were not clean at room temperature or at reflux; only product isolated was known 3-amino-2H-2-chromenone 14 (Scheme 6).21
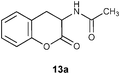 |
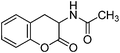
| | Fig. 2
N-Acetyl-ortho-tyrosine lactone
| |
We were pleased to find that reduction of the 4H-chromene 12a with Zn in a mixture of Ac2O and AcOH at 110 °C provided N-acetyl ortho-tyrosine lactone 13a in 68% yield (Scheme 7). Reduction with Fe and a mixture of AcOH and Ac2O was not clean.
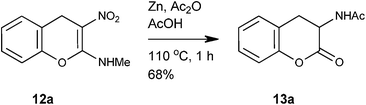 |
| | Scheme 7 Synthesis of N-acetyl-ortho-tyrosine lactone 13a. | |
We have identified four of the five possible phenolic hydroxyl protected lactones of N-acetyl-DOPA isomers 13b–e as targets for synthesis. Literature survey on DOPA isomers revealed only a few reports on their synthesis (Fig. 3).22,23 We employed Zn/AcOH/Ac2O as a general reducing reagent for the reduction of nitro group to acetyl amino with tandem hydrolysis of enamine functionality to lactone carbonyl in 4H-chromenes 12. Thus, reduction of the isomeric methoxy/benzyloxy-3-nitro-4H-2-chromenyl]-N-methylamines 12b–e provided corresponding lactones of four protected DOPA isomers 13b–e (Scheme 8; Table 2) in good yield.24 Overall, our method constitutes three-step high yielding synthesis of the lactones of the protected amino acids 13a–e from abundantly available NMSM 6 and 2-hydroxybenzaldehydes 5.
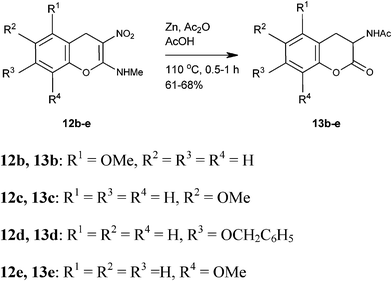 |
| | Scheme 8 Synthesis of four DOPA isomers 13b–e | |
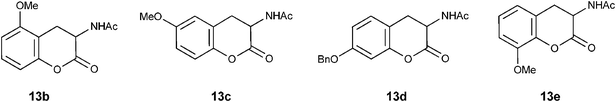 |
| | Fig. 3
Lactones of protected DOPA isomers | |
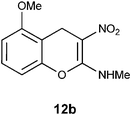
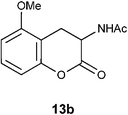
Table 2 Treatment of 12b–e with Zn, Ac2O and AcOH to get lactones of DOPA isomers 13b–e
| Entry |
Subsrate |
Product |
Time (h) |
Yield (%) |
| 1 |
|
|
1 |
68 |
| 2 |
|
|
1 |
82 |
| 3 |
|
|
0.5 |
61 |
| 4 |
|
|
1 |
68 |
Reaction of 2-methylamino-3-nitro-4H-chromenes with electron rich aromatic compounds
Our next task was to replace the labile C4-SMe in 7a with electron rich aromatic compounds for a facile synthesis of 4-aryl-4H-chromenes. 4-Aryl-4H-chromenes are potent apoptosis inducing agents.25 They have been evaluated for cytotoxic activity26 and as a consequence, for the development of anticancer drugs.27 Indeed, some of the 4-aryl-4H-chromenes were found to be highly active against human lung tumor xenograft28 and effectively inhibit tubulin polymerization.29
We reacted the 4H-chromene 7a with electron rich compounds like N,N-dimethylaniline, indole and pyrrole. In each case, corresponding 4-aryl-4H-chromenes 15–17 were obtained in excellent yield (Scheme 9). Regioselective electrophilic substitution on N,N-dimethylaniline, indole and pyrrole took place in expected manner. Unfortunately, substitution of SMe in 7a with comparatively less electron rich heterocycles like furan or thiophene did not take place.
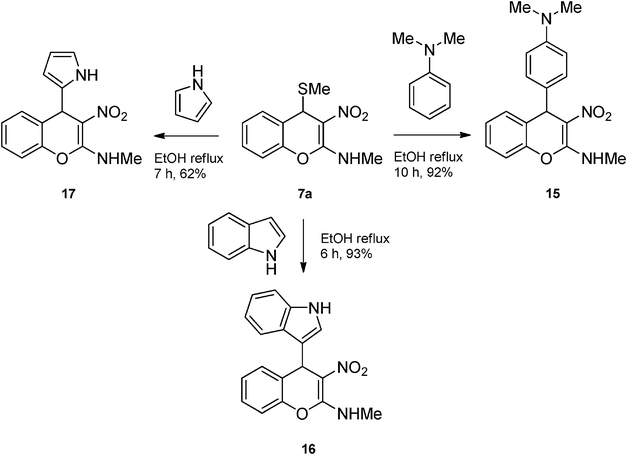 |
| | Scheme 9 Synthesis of 4-aryl-4H-chromenes 15-17 from 2-methylamino-3-nitro-4-methylthio-4H-chromene 7a and electron rich aromatic compounds. | |
Reactions of 4H-chromene 7a with nucleophilic reagents like alkyl amines (in EtOH), sodium azide (in acetone), sodium cyanide (in DMSO) or sodium methoxide (in MeOH) were also unsuccessful.
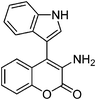
Synthesis of the lactone of tryptophan-orthotyrosine hybrid amino acid
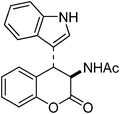
Our next undertaking was to convert 4-indolyl-4H-chromene 16 into the lactone of the hybrid amino acid 18 incorporating tryptophan and ortho-tyrosine units (Scheme 10). The Zn/AcOH/Ac2O reduction provided required lactone of the hybrid acetylated amino acid 18 in 65% yield. The 1H and 13C NMR spectra of the N-acetyl amino acid 18 displayed signals due to both indole as well as coumarin moieties. Interestingly, from this reaction, a single diastereomer of 18 was isolated, to which we assigned trans stereochemistry based upon the large coupling constant of 13.2 Hz between C3H and C4H. The Zn-AcOH reduction of 16 provided the enamine 19 in 32% yield.
Conclusions
In conclusion, we have demonstrated a facile reductive desulfurization of 2-alkyamino-3-nitro-4-alkylsulfanyl 4H-chromenes with Hantzsch diester to furnish 2-alkyamino-3-nitro-4H-chromenes. Reduction of the nitro group and release of the masked lactone in 2-alkyamino-3-nitro-4H-chromenes with Zn, Ac2O and AcOH lead to facile and high yielding synthesis of lactones of ortho-tyrosine, four isomers of DOPA. The C4 methylsulfanyl group in 4H-chromene 7a was readily replaced with electron rich aromatic compounds to provide C4 aryl substituted 4H-chromenes. Functional group transformation on 4-indolyl-4H-chromene provided the lactone of the hybrid amino acid incorporating tryptophan and ortho-tyrosine units. We are now concentrating on the substitution of C4-SMe in parent 4H-chromene 7a with phenols and their further transformation into hybrid amino acids.
Experimental
General methods
TLC was performed with silicagel-G or silica gel GF-254 using hexanes/ethyl acetate as eluent and the spots were visualized by short exposure to iodine vapor or UV light. Column chromatography was carried on silica gel (100–200 mesh) using increasing percentage of ethyl acetate in hexanes. The ultraviolet spectra were recorded on Hitachi U 1800 Ratio Beam spectrophotometer using MeOH as solvent and absorption wavelength (λmax) and molar absorbance (A) were recorded. The IR spectra were recorded as neat on a NaCl crystal or as solid-solution in KBr using ABB BOMEM MB 104 FT-IR spectrometer and only selected absorbencies (νmax) are reported. The 1H NMR spectra were recorded using BRUKER 300 or 400 MHz instruments in 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 CCl4-CDCl3 or in 9:1 CDCl3-DMSO-D6. Chemical shifts (δH) are quoted in parts per million (ppm), referenced to TMS. Each coupling constant is denoted as J and values are given in Hz. The 13C NMR spectra were recorded at 75 or 100 MHz on BRUKER 300 or 400 MHz instruments respectively. Chemical shifts (δC) are quoted in parts per million (ppm), referenced to TMS. The 1H NMR spectral features are designated as follows: m = multiplet, t = triplet, d = doublet, s = singlet and br = broad. HRMS were recorded using Micro-Mass ESI-TOF MS spectrometer. Melting points were determined in GallenKamp melting point apparatus and were uncorrected. The preparative procedures and the spectral data of the new compounds prepared in this study are described below.
:1 CCl4-CDCl3 or in 9:1 CDCl3-DMSO-D6. Chemical shifts (δH) are quoted in parts per million (ppm), referenced to TMS. Each coupling constant is denoted as J and values are given in Hz. The 13C NMR spectra were recorded at 75 or 100 MHz on BRUKER 300 or 400 MHz instruments respectively. Chemical shifts (δC) are quoted in parts per million (ppm), referenced to TMS. The 1H NMR spectral features are designated as follows: m = multiplet, t = triplet, d = doublet, s = singlet and br = broad. HRMS were recorded using Micro-Mass ESI-TOF MS spectrometer. Melting points were determined in GallenKamp melting point apparatus and were uncorrected. The preparative procedures and the spectral data of the new compounds prepared in this study are described below.
N2-Methyl-4-[2-(methylamino)-3-nitro-4H-4-chromenyl]-3-nitro-4H-2-chromenamine 9.
To a stirred suspension of Raney ® Ni (0.13 g, 2.2 mmol) in 8 mL of ethanol, (0.25 g, 0.99 mmol) of N-methyl-N-[4-(methylsulfanyl)-3-nitro-4H-2-chromenyl] amine 7a was added under nitrogen atmosphere and the resulting reaction mixture was refluxed for 15 h. Cooled reaction mixture was filtered through celite. Evaporation of the solvent and purification by column chromatography with increasing amounts of EtOAc (20% to 40%) in hexanes provided 0.04 g of 9 in 11% yield as a colorless solid; mp 152 °C; Rf = 0.6 (3:2, hexane-EtOAc); IR (KBr) 3441, 3199, 2944, 1644, 1609, 1465, 1401, 1347, 1217, 1172, 1061, 759 cm−1; 1H NMR (300 MHz, DMSO-D6:CDCl3) δH 10.27 (br s, 2H), 7.28 (t, J = 7.2 Hz, 2H), 7.11 (t, J = 7.2 Hz, 2H), 6.88 (d, J =8.0 Hz, 2H), 6.80 (d, J = 7.2 Hz, 2H), 4.82 (s, 2H), 2.87 & 2.86 (s, 6H); 13C NMR (75 MHz, DMSO-D6:CDCl3) δC 160.1 (C), 148.9 (C), 129.2 (CH), 128.8 (CH), 124.2 (CH), 120.4 (C), 115.0 (CH), 104.7 (C), 39.1 (CH), 27.5 (Me); HRMS (ESI+): calcd for C20H18N4O6 (MNa+), 433.1124; found, 433.1129.
N2-Methyl-2H-2,3-chromenediamine 10.
To a stirred suspension of Raney ® Ni (0.35 g, 5.9 mmol) in 8 mL of ethanol, 7a (0.2 g, 0.79 mmol) was added and stirred at room temperature for 2 h. After disappearance of 7a (TLC), the reaction mixture was filtered through celite. Evaporation of the solvent and separation through column chromatography with increasing amounts of EtOAc (20% to 30%) in hexanes provided 10 in 12% yield as colorless solid; mp 136 °C, Rf = 0.6 (7:3 hexane-EtOAc); IR (KBr) 3362, 2919, 1662, 1633, 1457, 1178, 1109, 748 cm−1; 1H NMR (300 MHz, CDCl3) δH 10.5 (br s, 1H), 7.07–7.0 (m, 4H), 6.1 (s, 1H), 4.21 (s, 2H), 3.2 (s, 3H); 13C NMR (75 MHz, CDCl3) δC 149.6 (C), 134.3 (C), 124.9 (CH), 124.4 (2 x CH), 123.2 (CH), 122.0 (C), 114.6 (CH), 102.1 (CH), 33.1 (Me); Anal. Calcd for C10H12N2O: C, 68.16; H, 6.86; N, 15.9. Found: C, 68.13; H, 6.89; N, 15.4.
Preparation of N-methyl-N-(3-nitro-4H-2-chromenyl)amine 12a: Method A: Using TBTH and AIBN.
To a solution of 7a (0.5 g, 2.0 mmol) in benzene (5 mL), tri-n-butyltin hydride (TBTH, 0.63 g, 2.1 mmol) and 2,2’-azabisisobutyronitrile (AIBN, 0.17 g, 1.05 mmol) were added and refluxed for 0.5 h in a preheated oil bath maintained at 95 °C. After completion of the reaction (TLC) the solvent was evaporated under reduced pressure. Remaining portion was extracted with DCM (25 mL), washed with water (30 ml × 2) and brine (20 mL). Organic layer was separated and dried over anhydrous sodium sulfate. Evaporation of the solvent and purification by column chromatography with increasing amounts of EtOAc (20% to 30%) in hexanes provided 0.34 g of 12a in 82% yield. mp >200 °C (EtOH); Rf = 0.6 (1:1 hexane-EtOAc); UV (MeOH) λmax 351 nm (log ε = 4.5), 269 nm (shoulder); IR (KBr) 3170, 1657, 1475, 1370, 1258, 1221, 1173, 1057, 757 cm−1; 1H NMR (300 MHz, CCl4:CDCl3) δH 10.64 (br s, 1H), 7.25 (d, J = 7.5 Hz, 1H), 7.2–7.1 (m, 2H), 7.06 (d, J = 8.1 Hz, 1H), 3.98 (s, 2H), 3.2 (d, J = 5.1 Hz, 3H); 13C NMR (75 MHz, CCl4:CDCl3 1:1) δC 159.4 (CH), 148.1 (C), 129.5 (CH), 128.1 (CH), 125.6 (CH), 120.1 (C), 116.1 (CH), 100.1 (C), 27.8 (NHMe), 25.6 (CH2); Anal. Calcd for C10H10N2O3: C, 58.25; H, 4.89; N, 13.59. Found: C, 58.27; H, 4.92; N, 13.63.
Method B: Using thiophenol.
To 7a (0.5 g, 2.0 mmol), benzenethiol (1.7 g, 10 equivalents) was added and the solution was refluxed for 2.5 h. The reaction mixture was diluted with ice-cold EtOH (3 mL) and the solid settled was filtered to provide 0.36 g of 12a in 88% yield.
Method C using Hantzsch diester (General procedure reductive removal of C4-SMe in 7).
To a mixture of 7a (0.5 g, 2.0 mmol) and diethyl 2,6-dimethyl-1,4-dihydro-3,5-pyridinedicarboxylate (Hantzsch diester, 0.501 g, 2.0 mmol) MeOH (10 mL) was added and the solution was refluxed for 4 h by which time the reaction was complete (TLC; hexanes:EtOAc, 1:1). The reaction mixture was allowed to cool to rt and the solvent was evaporated under reduced pressure. Purification by column chromatography with increasing amounts of EtOAc (20% to 30%) in hexanes provided 0.32 g of 12a in 78% yield as light yellow solid.
N-(5-Methoxy-3-nitro-4H-2-chromenyl)-N-methylamine 12b.
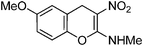
Following the general procedure (method C) described above, the reaction of 7b (0.5 g, 1.8 mmol) with diethyl 2,6-dimethyl-1,4-dihydro-3,5-pyridinedicarboxylate (0.45 g, 1.8 mmol) in MeOH (10 mL) reflux for 7 h provided 0.31 g of 12b in 74% yield as light yellow solid after column purification with 50% EtOAc in hexanes; mp 206–208 °C (EtOH); Rf = 0.4 (1:1 hexane-EtOAc); UV (MeOH) λmax 351 nm (log ε = 5.8), 288 nm (log ε = 5.2); IR (KBr) 3184, 2902, 1652, 1463, 1364, 1230, 1168, 1087, 1053, 749, 692 cm−1; 1H NMR (300 MHz, CDCl3) δH 10.72 (br s, 1H), 7.20 (t, J = 8.7 Hz, 1H), 6.69 (d, J = 8.7 Hz, 1H), 6.66 (d, J = 8.7 Hz, 1H), 3.86 (s, 3H), 3.81 (s, 2H), 3.21 & 3.14 (s, 3H); 13C NMR (75 MHz, CDCl3) δC 159.3 (C), 157.5 (C), 148.5 (C), 128.1 (CH), 109.4 (C), 108.0 (CH), 106.6 (CH), 104.6 (C), 55.8 (OMe), 27.8 (NHMe), 21.5 (CH2); HRMS (ESI+): calcd for C11H12N2O4 (MNa+), 259.0695; found, 259.0692. Anal. Calcd for C11H12N2O4: C, 55.93; H, 5.12; N, 11.86. Found: C, 55.88; H, 4.96; N, 11.79.
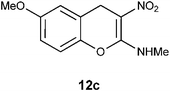
N-(6-Methoxy-3-nitro-4H-2-chromenyl)-N-methylamine 12c.
Following the general procedure (method C) described above, the reaction of 7c (1.0 g, 3.6 mmol) with diethyl 2,6-dimethyl-1,4-dihydro-3,5-pyridinedicarboxylate (0.90 g, 3.6 mmol) in MeOH (10 mL) reflux for 9 h provided 0.64 g of 12c in 76% yield as light yellow solid; mp 216–218 °C (EtOH); Rf = 0.4 (1:1 hexane-EtOAc); UV (MeOH) λmax 352 nm (log ε = 5.8), 266 nm (log ε = 5.2); IR (KBr) 3190, 2900, 1652, 1460, 1407, 1373, 1259, 1211, 1182, 1060, 727 cm−1; 1H NMR (300 MHz, CDCl3) δH 10.7 (br s, 1H), 7.02 (d, J = 8.7 Hz, 1H), 6.77 (d, J = 8.7 Hz, 1H), 6.73 (s, 1H), 3.97 (s, 3H), 3.80 (s, 2H), 3.2 (d, J = 5.4 Hz, 3H); 13C NMR (75 MHz, CDCl3) δC 159.6 (C), 157.0 (C), 142.1 (C), 120.8 (C), 117.0 (CH), 114.1 (CH), 113.2 (CH), 104.0 (C), 55.7 (OMe), 27.8 (NHMe), 26.0 (CH2); HRMS (ESI+): calcd for C11H12N2O4 (MNa+), 259.0695; found, 259.0692.
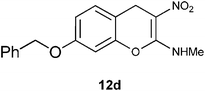
N-[7-(Benzyloxy)-3-nitro-4H-2-chromenyl]-N-methylamine 12d.
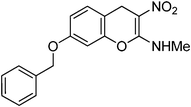
Following the general procedure (method C) described above, the reaction of 7d (1.0 g, 2.8 mmol) with diethyl 2,6-dimethyl-1,4-dihydro-3,5-pyridinedicarboxylate (0.71 g, 2.8 mmol) in MeOH (10 mL) reflux for 7 h provided 0.72 g of 12d in 82% yield as light yellow solid after column purification with 50% EtOAc in hexanes; mp 204–208 °C (EtOH); Rf = 0.4 (1:1 hexane-EtOAc); UV (MeOH) λmax 351 nm (log ε = 4.3), 288 nm (log ε = 3.7); IR (KBr) 3184, 2953, 1651, 1478, 1374, 1289, 1161, 1066, 749 cm−1; 1H NMR (400 MHz, CDCl3) δH 10.71 (br s, 1H), 7.41–7.14 (m, 6H), 6.82–6.7 (m, 2H), 5.06 (s, 2H), 3.91 (s, 2H), 3.2 (d, J = 5.2 Hz, 3H); 13C NMR (100 MHz, CDCl3) δC 159.4 (C), 158.5 (C), 148.4 (C), 136.3 (C), 130.0 (CH), 128.7 (2 x CH), 128.2 (CH), 127.4 (2 x CH), 112.9 (CH), 111.9 (C), 104.6 (C), 102.5 (CH), 70.4 (CH2), 27.9 (NHMe), 25.0 (CH2); HRMS (ESI+): calcd for C17H16N2O4 (MNa+), 335.3097; found, 335.3094.
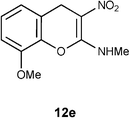
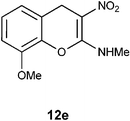
N-(8-Methoxy-3-nitro-4H-2-chromenyl)-N-methylamine 12e.
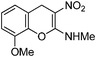
Following the general procedure (method C) described above, the reaction of 7e (0.5 g, 1.8 mmol) with diethyl 2,6-dimethyl-1,4-dihydro-3,5-pyridinedicarboxylate (0.45 g, 1.8 mmol) in MeOH (10 mL) reflux for 8 h provided 0.30 g of 12e in 72% yield as light yellow solid after recrystallization with MeOH (10 mL); mp 232 °C (EtOH); Rf = 0.4 (1:1 hexane-EtOAc); UV (MeOH): λmax 352 nm (log ε = 4.5), 290 nm (log ε = 3.9); IR (KBr) 3162, 2951, 1657, 1471, 1373, 1204, 1176, 1091, 773, 683 cm−1; 1H NMR (400 MHz, DMSO-D6) δH 10.45 (br s, 1H) 7.1 (t, J = 8.0 Hz, 1H), 6.99–7.01 (m, 1H), 6.88–6.90 (m, 1H), 3.85 (s, 3H), 3.09 (d, J = 4.8 2H); 13C NMR (400 MHz, DMSO-D6) δC 158.3 (C), 147.2 (C), 137.1 (C), 125.2 (C), 120.9 (CH), 120.2 (CH), 111.0 (CH), 102.9 (C), 56.1 (OMe), 27.9 (NHMe), 25.3 (CH2) ; HRMS (ESI+): calcd for C11H12N2O4 (MNa+), 259.0695; found, 259.0694.
Representative procedure for the reduction of nitro group with tandem hydrolysis of enamino group. Preparation of N1-(2-oxo-3,4-dihydro-2H-3-chromenyl)acetamide 13a.
To a well stirred solution of 12a (0.5 g, 2.4 mmol) in Ac2O (14.1 g, 13.0 mL, 138 mmol), AcOH (1.46 g, 1.4 mL, 24.2 mmol) was added and heated in a preheated oil bath maintained at 110 °C for 15 min. Zinc powder (3.2 g, 48.5 mmol) was added at the same temperature in portion wise over 5 min. The reaction mixture was vigorously stirred at this temperature for 1 h then allowed to cool to room temperature then filtered through celite. The filterate was diluted with DCM (30 mL), water (25 mL), brine solution (20 mL) and dried over anhydrous Na2SO4. Evaporation of the solvent and separation by column chromatography with increasing amounts of hexanes (20% to 80%) in EtOAc provided 0.31 g of 13a in 62% yield as colorless solid; mp 158 °C (EtOH); Rf = 0.4 (2:8 hexane-EtOAc); IR (KBr) 3920, 1762, 1635, 1542, 1373, 1234, 1161, 756 cm−1; 1H NMR (300 MHz, CDCl3) δH 7.29 (br t, J = 8.1 Hz, 1H), 7.21 (d, J = 7.5 Hz, 1H), 7.14 (t, J = 7.5 Hz, 1H), 7.05 (d, J = 8.1 Hz, 1H), 6.61 (br s, 1H), 4.78 (dt, J = 13.5 Hz, 6.6 Hz, 1H), 3.49 (dd, J = 15.0 Hz, 6.6 Hz, 1H), 2.92 (t, J = 15.0 Hz, 1H), 2.1 (s, 3H); 13C NMR (75 MHz, CDCl3) δC 170.2 (C), 168.6 (C), 150.7 (C), 128.8 (CH), 128.7 (CH), 125.0 (CH), 121.3 (C), 116.6 (CH), 48.3 (CH), 30.3 (CH2), 23.1 (CH3); HRMS (ESI+): calcd for C11H11NO3 (MNa+), 228.0637; Found, 228.0632.
N1-(5-Methoxy-2-oxo-3,4-dihydro-2H-3-chromenyl)acetamide 13b.
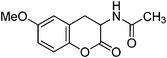
Following the procedure described for preparation of 13a, the reduction of 12b (0.25 g, 1.1 mmol) with zinc powder in the presence of Ac2O (6.2 g, 5.7 mL, 60 mmol) and AcOH (0.65 g, 0.6 mL, 10.5 mmol) on 1 h heating at 110 °C provided 0.17 g of 13b in 68% yield as colorless solid; mp 166–168 °C (EtOH); Rf = 0.3 (1:1 hexane-EtOAc); UV (MeOH) λmax 365 nm (log ε = 4.3), 284 nm (log ε = 4.1), 234 nm (log ε = 4.5); IR (KBr) 3384, 3284, 1753, 1657, 1594, 1466, 1372, 1268, 1236, 1080, 774 cm−1; 1H NMR (300 MHz, CDCl3) δH 7.22 (t, J = 8.7 Hz, 1H), 6.69 (d, J = 8.7 Hz, 1H), 6.68 (d, J = 8.7 Hz, 1H), 6.43 (br s, 1H), 4.75 (dt, J = 13.5 Hz, 6.6 Hz, 1H), 3.84 (s, 3H), 3.79 (dd, J = 15.9 Hz, 6.9 Hz, 1H), 2.53 (dd, J = 15.3 Hz, 14.1 Hz, 1H), 2.11 (s, 3H); 13C NMR (75 MHz, CDCl3) δC 170.1 (C), 168.7 (C), 157.0 (C), 151.6 (C), 128.9 (CH), 110.0 (C), 109.0 (CH), 106.8 (CH), 55.8 (CH), 48.0 (OMe), 24.7 (CH3), 23.1 (CH2); HRMS (ESI+): calcd for C12H13NO4 (MNa+), 258.0743; Found, 258.0739.
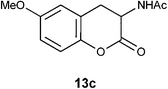
N1-(6-Methoxy-2-oxo-3,4-dihydro-2H-3-chromenyl)acetamide 13c.
Following the procedure described for preparation of 13a, the reaction of 12c (0.25 g, 1.1 mmol) with zinc powder (1.38 g, 21 mmol) in the presence of Ac2O (6.2 g, 5.7 mL, 60 mmol) and AcOH (0.65 g, 0.6 mL, 10.5 mmol) on 1 h heating at 110 °C provided 0.15 g of 13c in 62% yield as colorless solid after recrystallization using MeOH (3 mL); mp 194 °C (EtOH); Rf = 0.3 (1:1 hexane-EtOAc); UV (MeOH) λmax 353 nm (log ε = 4.3), 284 nm (log ε = 3.7); IR (KBr) 3298, 1749, 1637, 1541, 1494, 1373, 1201, 1035, 671 cm−1; 1H NMR (300 MHz, CDCl3) δH 6.94 (d, J = 9.0 Hz, 1H), 6.76 (dd, J = 8.7 Hz, 2.4 Hz, 1H), 6.68 (s, 1H), 6.61 (br s, 1H), 4.70 (dt, J = 14.1 Hz, 6.3 Hz, 1H), 3.74 (s, 3H), 3.39 (dd, J = 15.1 Hz, 6.6 Hz, 1H), 2.86 (dd, J = 14.7 Hz, 14.1 Hz, 1H), 2.07 (s, 3H); 13C NMR (75 MHz, CDCl3) δC 170.2 (C), 168.7 (C), 156.6 (C), 144.6 (C), 122.3 (CH), 117.5 (C), 114.1 (CH), 113.4 (CH), 55.7 (CH), 48.3 (OMe), 30.6 (CH2), 23.1 (CH3); HRMS (ESI+): calcd for C12H13NO4 (MNa+), 258.0743; found, 258.0742.
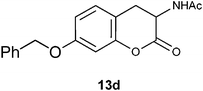
N1-[7-(Benzyloxy)-2-oxo-3,4-dihydro-2H-3-chromenyl]acetamide 13d.
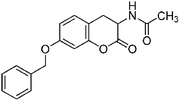
Following the procedure described for preparation of 13a, the reaction of 12d (0.5 g, 1.6 mmol) with zinc powder (2.1 g, 32 mmol) in the presence of Ac2O (9.3 g, 8.6 mL, 90 mmol) and AcOH (1.0 g, 0.9 mL, 16 mmol) on 0.5 h heating at 110 °C furnished 0.30 g of 13d in 61% yield as colorless solid after column purification with increasing amounts of EtOAc (20% to 50%) in hexanes; mp 150 °C (EtOH); Rf = 0.3 (1:1 hexane-EtOAc); UV (MeOH) λmax 354 nm (log ε = 4.6), 284 nm (log ε = 4.1); IR (KBr) 3302, 2904, 1757, 1644, 1536, 1374, 1271, 1156, 1032, 737, 694 cm−1; 1H NMR (300 MHz, CDCl3) δH 7.43–7.33 (m, 5H), 7.10 (d, J = 8.1 Hz, 1H), 6.76 (dd, J = 8.4 Hz, 2.4 Hz, 1H), 6.69 (d, J = 2.4 Hz, 1H), 6.41 (br s, 1H), 5.05 (s, 2H), 4.73 (dt, J = 13.5 Hz, 6.3 Hz, 1H), 3.46 (dd, J = 15.0 Hz, 6.6 Hz, 1H), 2.81 (dd, J = 14.1 Hz, 14.1 Hz, 1H), 2.10 (s, 3H); 13C NMR (75 MHz, CDCl3) δC 170.2 (C), 168.6 (C), 159.2 (C), 151.4 (C), 136.3 (C), 129.2 (CH), 128.7 (2 × CH), 128.2 (CH), 127.4 (2 × CH), 113.3 (C), 112.0 (CH), 103.5 (CH), 70.4 (CH2Ph), 48.7 (CH), 29.8 (CH2), 23.1 (CH3); HRMS (ESI+): calcd for C18H17NO4 (MNa+), 334.1056; found, 334.1054.
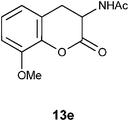
N1-(8-Methoxy-2-oxo-3,4-dihydro-2H-3-chromenyl)acetamide 13e.
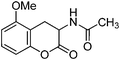
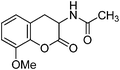
Following the procedure described for preparation of 13a, the reaction of 12e (0.5 g, 2.1 mmol) with zinc dust (2.8 g, 42 mmol) in the presence of Ac2O (12.3 g, 11.4 mL, 120 mmol) and AcOH (1.3 g, 1.2 mL, 21 mmol) on 1 h heating at 110 °C provided 0.34 g of 13e in 68% yield as colorless solid after recrystallization using MeOH (5 mL); mp 138–140 °C (EtOH); Rf = 0.3 (1:1 hexane-EtOAc); IR (KBr) 3315, 2900, 1762, 1644, 1540, 1485, 1324, 1276, 1174, 1093, 687 cm−1; 1H NMR (300 MHz, CDCl3) δH 7.72 (d, J = 7.2 Hz, 1H), 7.07 (t, J = 8.1 Hz, 1H), 6.88 (d, J = 8.1 Hz, 1H), 6.76 (d, J = 7.2 Hz, 1H), 4.85 (dt, J = 14.4 Hz, 6.9 Hz, 1H), 3.87 (s, 3H), 3.25 (dd, J = 15.6 Hz, 6.6 Hz, 1H), 3.04 (dd, J = 14.4 Hz, 15.0 Hz, 1H), 2.05 (s, 3H); 13C NMR (75 MHz, CDCl3) δC 170.0 (C), 167.5 (C), 147.0 (C), 139.7 (C), 124.5 (CH), 122.4 (CH), 119.5 (C), 111.0 (CH), 55.6 (CH), 47.2 (OMe), 30.1 (CH2), 22.5 (CH3); HRMS (ESI+): calcd for C12H13NO4 (MNa+), 258.0743; found, 258.0745.
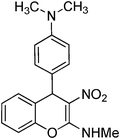
Representative procedure for the substitution of C4-SMe with electron rich aryl rings. Preparation of N2-methyl-4-[4-(dimethylamino)phenyl]-3-nitro-4H-2-chromenamine 15.
To a solution of 7a (0.5 g, 2.0 mmol) in ethanol (15 mL) N,N-dimethyl-N-phenylamine (0.26 g, 2.2 mmol) was added and reaction mixture was refluxed for 10 h by which time the reaction was complete (TLC). The reaction mixture was cooled to room temperature and kept aside for 3 h. The compound crystallized out was separated by filtration. The solid was washed with ice-cold ethanol (5 mL) to yield 0.59 g of 15 in 92% yield as a colorless solid; mp 196–199 °C (EtOH); Rf = 0.4 (6:4 hexane-EtOAc); UV (MeOH) λmax 349 nm (log ε = 4.3), 263 nm (log ε = 4.4); IR (KBr) 2888, 2801, 1638, 1609, 1521, 1469, 1402, 1356, 1210, 1133, 1062, 942, 811, 761, 692 cm−1; 1H NMR (300 MHz, CDCl3 + DMSO-D6) δH 10.3 (br s, 1H), 7.54–7.06 (m, 6H), 6.58 (d, J = 8.4 Hz, 2H), 5.29 (s, 1H), 3.24 (d, J = 5.1 Hz, 3H), 2.85 (s, 6H); 13C NMR (75 MHz, CDCl3:DMSO-D6) δC 158.7 (C), 148.7 (C), 146.8 (C), 131.1 (2 × C), 128.7 (CH), 127.4 (2 × CH), 127.1 (CH), 125.0 (CH), 115.3 (CH), 111.7 (2 × CH), 108.4 (C), 40.6 (CH), 40.3 (CH), 39.8 (CH3), 27.2 (Me); Anal. Calcd for C18H19N3O3: C, 66.45; H, 5.89; N, 12.91; Found: C, 66.38; H, 5.91; N, 12.87.
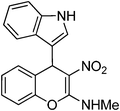
N-[4-(1H-3-indolyl)-3-nitro-4H-2-chromenyl]-N-methylamine 16.
Following the procedure described for 15, the reaction of 7a (0.5 g, 2.0 mmol) with 1H-indole (0.23 g, 2.2 mmol) in ethanol (15 mL) on 6 h reflux provided 0.59 g of 16 in 93% yield as colorless solid; mp >200 °C (EtOH); Rf = 0.4 (1:1 hexane-EtOAc); UV (MeOH) λmax 350 nm (log ε = 4.1), 272 nm (log ε = 3.9); IR (KBr) 3422, 1638, 1607, 1546, 1466, 1362, 1237, 1206, 1167, 1059, 797, 736, 692, 610 cm−1; 1H NMR (400 MHz, DMSO-D6) δH 10.90 (br s, 1H), 10.34 (d, J = 4.4 Hz, 1H), 7.38–7.22 (m, 6H), 7.11 (t, J = 7.2 Hz, 1H), 6.98 (t, J = 7.2 Hz, 1H), 6.88 (t, J = 7.2 Hz, 1H), 5.65 (s, 1H), 3.21 (d, J = 4.4 Hz, 3H); 13C NMR (100 MHz, DMSO-D6) δC 159.0 (C), 147.5 (C), 136.4 (C), 129.3 (C), 127.9 (CH), 125.6 (CH), 125.4 (CH), 125.2 (CH), 123.2 (C), 120.9 (CH), 118.7 (CH), 118.0 (CH), 117.2 (CH), 116.0 (CH), 111.7 (C), 107.6 (C), 33.2 (CH), 28.1 (Me); Anal. Calcd for C18H15N3O3: C, 67.28; H, 4.71; N, 13.08; Found: C, 67.35; H, 4.61; N, 13.01.
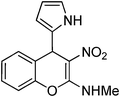
N-Methyl-N-[3-nitro-4-(1H-2-pyrrolyl)-4H-2-chromenyl]amine 17.
Following the procedure described for 15, the reaction of 7a (0.5 g, 2.0 mmol) with pyrrole (0.398 g, 5.9 mmol) in ethanol (15 mL) for 7 h reflux provided 0.36 g of 17 in 67% yield as colorless solid; mp 199 °C (EtOH); Rf = 0.4 (1:1 hexane-EtOAc); UV (MeOH) λmax 352 nm (log ε = 4.5), 269 nm (log ε = 4.1); IR (KBr) 3378, 3209, 1638, 1606, 1462, 1393, 1337, 1243, 1168, 1062, 761, 730 cm−1; 1H NMR (400 MHz, CDCl3) δH 10.56 (br s, 1H), 9.24 (br s, 1H), 7.20–7.15 (m, 4H), 6.68 (s, 1H), 5.97 (br s, 1H), 5.72 (s, 1H), 5.20 (s, 1H), 3.19 (d, J = 5.1 Hz, 3H); 13C NMR (100 MHz, CDCl3) δC 160.1 (C), 148.6 (C), 134.5 (C), 130.2 (CH), 128.7 (CH), 125.8 (CH), 122.5 (C), 117.4 (CH), 115.9 (CH), 109.3 (C), 107.2 (CH), 105.6 (CH), 34.3 (CH), 27.9 (Me). HRMS (ESI+): calcd for C14H13N3O3 (MNa+), 294.0854; found, 294.0854.
N1-[4-(1H-3-Indolyl)-2-oxo-3,4-dihydro-2H-3-chromenyl]acetamide 18.
By following the procedure described for 13a, the reaction of 16 (0.5 g, 1.6 mmol) with zinc powder (2.0 g, 31 mmol) in the presence of Ac2O (9.0 g, 8.3 mL, 88 mmol) and AcOH (1.0 g, 0.9 mL, 16 mmol) on heating at 110 °C for 1 h furnished 0.32 g of 18 in 65% yield as colorless solid; mp 134–138 °C (EtOH); Rf = 0.5 (2:8 hexane-EtOAc); IR (KBr) 3388, 3068, 1762, 1658, 1542, 1456, 1373, 1334, 1141, 752 cm−1; 1H NMR (300 MHz, CDCl3) δH 8.7 (br s, 1H), 7.4–6.9 (m, 8H), 6.81 (d, J = 7.5 Hz, 1H), 6.25 (d, J = 11.1 Hz, 1H), 5.25 (dd, J = 13.2 Hz, 8.7 Hz, 1H), 4.77 (d, J = 13.2 Hz, 1H), 1.76 (s, 3H); 13C NMR (75 MHz, CDCl3) δC 170.3 (C), 167.4 (C), 149.6 (C), 136.0 (2C), 128.4 & 128.1 (CH), 126.2 (C), 125.2 (CH), 124.4 (CH), 123.3 (CH), 121.9 (CH), 119.4 (CH), 118.3 (CH), 116.1 (CH), 111.2 (CH) 110.0 (C) , 51.6 (CHNH), 37.3 (CH), 22.3 (CH3). Anal. Calcd for C19H16N2O3: C, 71.24; H, 5.03; N, 8.74; Found: C, 71.31; H, 5.12; N, 8.67.
3-Amino-4-(1H-3-indolyl)-2H-2-chromenone 19.
Following the representative procedure described above the reaction of 16 (0.5 g, 1.6 mmol) with zinc powder (1.46 g, 0.016 mmol) in AcOH (9.4 g, 9 mL) medium at 110 °C provided 0.14 g of 19 in 32% as brown color solid after column purification with increasing amounts of EtOAc (20% to 50%) in hexanes; mp >200 °C (EtOH); Rf = 0.6 (1:1 hexane-EtOAc); UV (MeOH) λmax 334 nm (log ε = 4.0), 275 nm (log ε = 4.0), 252 nm (log ε = 4.0); IR (KBr) 3435, 3341, 3299, 1679, 1619, 1553, 1452, 1429, 1325, 1296, 1185, 1114, 755 cm−1; 1H NMR (300 MHz, CDCl3: DMSO-D6) δH 11.01 (br s, 1H), 7.55 (d, J = 2.4 Hz, 1H), 7.4–7.0 (m, 8H), 4.47 (br s, 2H); 13C NMR (75 MHz, CDCl3) δC 158.5 (C), 147.7 (C), 136.0 (C), 129.0 (C), 125.6 (CH), 124.9 (C), 124.6 (CH), 124.3 (CH), 123.5 (CH), 121.6 (C), 121.5 (CH), 119.7 (CH), 119.2 (CH), 115.7 (C), 115.3 (CH), 111.4 (CH), 105.7 (C); HRMS (ESI+): calcd for C17H12N2O2 (MH+), 277.0978, (MNa+), 299.0797; Found, (MH+), 277.0977, (MNa+), 299.0795.
Acknowledgements
H. S. P. R thanks UGC, UGC-SAP, CSIR and DST-FIST for financial assistance. K. G. and M. K. thank CSIR and PU respectively for fellowship. We thank IACS, Kolkata, and SAIC, IIT, Chennai for recording spectra. Shasun Chemicals and Drugs Ltd, Chennai gave the generous gift of NMSM.
References
-
(a) K. Ito, H. Ozasa, Y. Noda, Y. Koike, S. Arii and S. Horikaw, Clin. Nutr., 2008, 27, 773–780 CrossRef CAS;
(b) M.I. Aguilar, A. W. Purcell, R. Devi, R. Lew, J. Rossjohn, A. I. Smith and P. Perlmutter, Org. Biomol. Chem., 2007, 5, 2884–2890 RSC;
(c) E. Toth and A. Lajtha, Neurochem. Res., 1981, 6, 1309–1317 CrossRef CAS.
- G. A. Molnar, G. Wagner, L. Marko, T. Koszegi, M. Mohas, B. Kocsis, Z. Matus, L. Wagner, M. Tamasko, I. Mazak, B. Laczy, J. Nagy and I. Wittmann, Kidney Int., 2005, 68, 2281–2287 CrossRef CAS.
- T. Ishikawa, Heterocycles, 2000, 53, 453–474 CrossRef CAS.
- G. A. Molnar, V. Nemes, Z. Biro, A. Ludany, Z. Wagnar and I. Wittmann, Free Radical Res., 2005, 39, 1359–1366 CrossRef CAS.
- T. G. Huggins, M. C. W. Knecht, N. A. Detorie, J. W. Bayness and S. R. Thorpe, J. Biol. Chem., 1993, 268, 12341–12347 CAS.
-
(a) M. Makota, N. Taeko, K. Tomomi, I. Hitoshi, T. Masatake and S. Yukio, J. Health Sci., 2002, 48, 79–82 CrossRef;
(b) C. Krach, G. Sontag and S. Solar, Food Res. Int., 1999, 32, 43–47 CrossRef CAS;
(c) M. Werner, B. Renate and F. Dieter, Radiat. Phys. Chem., 1990, 35, 332–336 CrossRef.
- Y. Misu, Y. Goshima and T. Miyamae, Trends Pharmacol. Sci., 2002, 23, 262–268 CrossRef CAS.
-
R. Vardanyan and V. Hruby, Synthesis of Essential drugs,Elsevier, 2006 Search PubMed.
- A. Hutinee and A. Ziogas, Amino Acids, 1996, 11, 345–366 CrossRef.
-
(a) T. Hohsaka, Seikagaku, 2007, 79, 247–253 (
Chem. Abstr.
, 2007
, 146
, 496047
) CAS;
(b) C. H. Roehrig, O. A. Retz, L. Hareng, T. Hartung and R. R. Schmidt, ChemBioChem, 2005, 6, 1805–1816 (
Chem. Abstr.
, 2005
, 145
, 505752
) CrossRef CAS.
- H. S. P. Rao and K. Geetha, Tetrahedron Lett., 2009, 50, 3836–3839 CrossRef CAS.
- E. J. Corey, B. W. Erickson and R. Noyori, J. Am. Chem. Soc., 1971, 93, 1724–1729 CrossRef CAS.
- D. Dauzonne and R. Royer, Synthesis, 1983, 10, 836–837 CrossRef.
- E. Vedejs and W. Powell, J. Am. Chem. Soc., 1982, 104, 2046–2048 CrossRef CAS.
- D. Hedstrand, W. Kruizinga and R. Kellogg, Tetrahedron Lett., 1978, 19, 1255–1258 CrossRef.
-
(a) D. M. Stout, Chem. Rev., 1982, 82, 223–243 CrossRef CAS;
(b) X. Q. Zhu, H. Y. Wang, J. S. Wang and Y. C. Liu, J. Org. Chem., 2001, 66, 344–347 CrossRef CAS.
-
(a) For previous syntheses of ortho-tyrosine see: T. Matsuura, K. Matsui and K. Ichihara, Med. J. Osaka Univ., 1954, 4, 449–455 (
Chem. Abstr.
, 1955
, 49
, 6222
) CAS;
(b) C. Dugave, J. Org. Chem., 1995, 60, 601–607 CrossRef CAS;
(c) M. Ali, N. H. Khan and A. A. Siddiqui, Synth. Commun., 1976, 6, 227–235 CrossRef CAS;
(d) G. Kokotos and C. Tzougraki, J. Heterocycl. Chem., 1986, 23, 87–92 CrossRef CAS;
(e) C. Petitclerc, A. D’Iorio and N. L. Benoiton, J. Labelled Compd., 1969, 5, 265–269 CrossRef CAS.
- J. P. Nicolle, J. F. Hamon and M. Wakselman, Bull. Soc. Chim. Fr., 1977, 83–88 (
Chem. Abstr.
, 1977
, 87
, 151958
) CAS.
- S. G. Manjunatha, P. Chittari and S. Rajappa, Helv. Chim. Acta, 1991, 74, 1071–1080 CrossRef CAS.
- D. Basavaiah, R. M. Reddy, N. Kumaragurubaran and D. S. Sharada, Tetrahedron, 2002, 58, 3693–3697 CrossRef CAS.
- A. A. Kudale, J. Kendall, C. C. Warford, N. D. Wilkins and G. J. Bodwell, Tetrahedron Lett., 2007, 48, 5077–5080 CrossRef CAS.
-
(a) K. Hirai, Biochem. J., 1927, 189, 88–91 (
Chem. Abstr.
, 1928
, 22
, 37666
) CAS;
(b) V. G. Yokovlev, Dokl. Akad. Nauk SSSR, 1950, 71, 497–500 (
Chem. Abstr.
, 1950
, 44
, 40735
) Search PubMed;
(c) W. Ried and H. G. Gebhardtsbauer, Chem. Ber., 1956, 89, 2933–2939 (
Chem. Abstr.
, 1957
, 51
, 46915
) CrossRef CAS;
(d) D. F. Reinhold, J. H. Markillie and M. Sletzinger, Ger. Pat., 1964, 1183097 (
Chem. Abstr.
, 1965
, 62
, 10509f
) Search PubMed;
(e) T. Ueno, H. Fukami, H. Ohkishi, H. Kumagai and H. Yamada, Biochim. Biophys. Acta., 1970, 206, 476–479 CAS.
-
(a) J. P. Lambooy, J. Am. Chem. Soc., 1949, 71, 3758–3759 CrossRef CAS;
(b) J. P. Lambooy, J. Am. Chem. Soc., 1954, 76, 133–138 CrossRef CAS.
-
(a)
Y. Misu and Y. Goshima, Neurobiology of DOPA as neurotransmitter; CRC Press LLC: USA, 2006, p. 380 Search PubMed;
(b) Y. Misu, T. Kubo, K. Honjo and Y. Goshima, Central Mechanisms of Cardiovascular Regulation, 2007, 107–138 CAS;
(c) D. J. Brooks, Neuropsychiatric Disease and Treatment, 2008, 4, 39–47 CAS.
-
(a) W. Kemnitzer, S. Jiang, Y. Wang, S. Kasibhatla, C. Crogan-Grundy, M. Bubenik, D. Labrecque, R. Denis, S. Lamothe, G. Attardo, H. Gourdeau, B. Tseng and S. X. Cai, Bioorg. Med. Chem. Lett., 2008, 18, 603–607 CrossRef CAS;
(b) S. X. Cai, Recent Pat. Anticancer Drug Discov., 2007, 2, 79–101 CrossRef CAS;
(c) S. X. Cai, J. Drewe and S. Kasibhatla, Curr. Med. Chem., 2006, 13, 2627–2644 CrossRef CAS;
(d) D. Gree, S. Vorin, V. L. Manthati, F. Caijo, G. Viault, F. Manero, P. Juin and R. Gree, Tetrahedron Lett., 2008, 49, 3276–3278 CrossRef CAS;
(e) S. Sciabola, E. Carosati, L. C. Sanchez, M. Baroni and R. Mannhold, Bioorg. Med. Chem., 2007, 15, 6450–6462 CrossRef CAS;
(f) A. Afantitis, G. Melagraki, H. Sarimveis, P. A. Koutentis, J. Markopoulos and O. I. Markopoulou, Bioorg. Med. Chem., 2006, 14, 6686–6694 CrossRef CAS.
-
(a) J. M. Doshi, D. Tian and C. Xing, J. Med. Chem., 2006, 49, 7731–7739 CrossRef CAS;
(b) J. M. Doshi, D. Tian and C. Xing, Mol. Pharmaceutics, 2007, 4, 919–928 CrossRef CAS.
- S. Kasibhatla, H. Gourdeau, K. Meerovitch, J. Drewe, S. Reddy, L. Qiu, H. Zhang, F. Bergeron, D. Bouffard, Q. Yang, J. Herich, S. Lamothe, S. X. Cai and B. Tseng, Mol. Cancer Ther., 2004, 3, 1365–1373 CAS.
- H. Gourdeau, L. Leblond, B. Hamelin, C. Desputeau, K. Dong, I. Kianicka, D. Custeau, C. Boudreau, L. Geerts, S. X. Cai, J. Drewe, D. Labrecque, S. Kasibhatla and B. Tseng, Mol. Cancer Ther., 2004, 3, 1375–1384 CAS.
- W. Kemnitzer, J. Drewe, S. Jiang, H. Zhang, Y. Wang, J. Zhao, S. Jia, J. Herich, D. Labrecque, R. Storer, K. Meerovitch, D. Bouffard, R. Rej, R. Denis, C. Blais, S. Lamothe, G. Attardo, H. Gourdeau, B. Tseng, S. Kasibhatla and S. X. Cai, J. Med. Chem., 2004, 47, 6299–6310 CrossRef CAS.
Footnote |
| † Electronic Supplementary Information (ESI) available. See DOI: 10.1039/c1ra00461a |
|
| This journal is © The Royal Society of Chemistry 2011 |
Click here to see how this site uses Cookies. View our privacy policy here. 
![Synthesis hybrid amino acid 18 and 3-amino-indolyl chromenone 19 from N-[4-(1H-3-indolyl)-3-nitro-4H-2-chromenyl]-N-methylamine 16.](/image/article/2011/RA/c1ra00461a/c1ra00461a-s10.gif)