Borrelidin modulates the alternative splicing of VEGF in favour of anti-angiogenic isoforms†
J.
Woolard
b,
W.
Vousden
a,
S. J.
Moss
a,
A.
Krishnakumar
b,
M. V. R.
Gammons
b,
D. G.
Nowak
b,
N.
Dixon
d,
J.
Micklefield
d,
A.
Spannhoff
e,
M. T.
Bedford
e,
M. A.
Gregory
a,
C. J.
Martin
a,
P. F.
Leadlay
c,
M. Q.
Zhang
a,
S. J.
Harper
b,
D. O.
Bates
*b and
B.
Wilkinson
*a
aBiotica, Chesterford Research Park, Cambridge, CB10 1XL, UK. E-mail: barrie.wilkinson@biotica.com; Fax: +44 (0)1799 532921; Tel: +44 (0)1799 532925
bMicrovascular Research Laboratories, Bristol Heart Institute, Department of Physiology and Pharmacology, School of Veterinary Sciences, University of Bristol, Southwell Street Bristol, BS2 8EJ, UK. E-mail: dave.bates@bristol.ac.uk; Fax: +44 (0)117 9288151; Tel: +44 (0)117 9289818
cDepartment of Biochemistry, University of Cambridge, 80 Tennis Court Road, Cambridge, CB2 1GA, UK
dSchool of Chemistry and Manchester Interdisciplinary Biocentre, University of Manchester, 131 Princess Street, Manchester, M1 7DN, UK
eThe University of Texas M.D. Anderson Cancer Centre, Science Park-Research Division, Smithville, Texas 78957, USA
First published on 23rd September 2010
Abstract
The polyketide natural product borrelidin 1 is a potent inhibitor of angiogenesis and spontaneous metastasis. Affinity biopanning of a phage display library of colon tumour cell cDNAs identified the tandem WW domains of spliceosome-associated protein formin binding protein 21 (FBP21) as a novel molecular target of borrelidin, suggesting that borrelidin may act as a modulator of alternative splicing. In support of this idea, 1, and its more selective analog 2, bound to purified recombinant WW domains of FBP21. They also altered the ratio of vascular endothelial growth factor (VEGF) isoforms in retinal pigmented endothelial (RPE) cells in favour of anti-angiogenic isoforms. Transfection of RPE cells with FBP21 altered the ratio in favour of pro-angiogenic VEGF isoforms, an effect inhibited by 2. These data implicate FBP21 in the regulation of alternative splicing and suggest the potential of borrelidin analogs as tools to deconvolute key steps of spliceosome function.
Introduction
We previously reported the mutasynthesis1 of new analogs of the anti-bacterial2 and anti-angiogenic3,4polyketide borrelidin 1 using genetically engineered strains of the producing organism Streptomyces parvulus Tü4055. Of particular note was BC194 2 which exhibits reduced cytotoxic effects and a significantly enhanced ability (pM rather than nM) to inhibit in vitro models of angiogenesis.62 differs from 1 only in the cyclopentanecarboxylic acid moiety which is truncated to a cyclobutanecarboxylic acid. Modulation of the in vitro toxicity to angiogenesis inhibition profile of 1 was also reported elsewhere for analogs where the carboxylic acid moiety was modified synthetically.7 Taken together these data suggest that modifications in this region confer selectivity. 1 exhibits additional biological activities of interest including potent inhibition of spontaneous metastasis in vivo,4 and of cell migrationin vitro.8The antibacterial action of 1 is exerted uniquely through inhibition of threonyl-tRNA synthetase9 (ThrRS) but several poorly-understood mechanisms appear to be involved in its anti-angiogenic activity.3,5,10 It is probable that 1 inhibits mammalian ThrRS11 but this has not been verified at the biochemical level. In this article we report our efforts to identify the putative molecular target of 1 responsible for its potent anti-angiogenic activity and subsequent biological evaluation.
Results and discussion
To help clarify the molecular target(s) responsible for its anti-angiogenic activity, we coupled 1 and its biosynthetically-engineered1 analog 3, which is inactive in in vitro assays of angiogenesis5 and cell proliferation1,5 inhibition, to biotin in order to enable immobilization on streptavadin coated surfaces. This was achieved by activation of the carboxylic acid as a mixed-anhydride (treatment with isobutylchloroformate and dimethylaminopyridine), followed by reaction with biotinamidohexanoic acid hydrazide to yield 4 (from 1) and 5 (from 3) (see Fig. 1 and ESI†). The use of 3 provides a negative control against which to compare 1. As modification of the carboxylic acid moiety enhances selectivity in vitro, we anticipated an enrichment for angiogenesis inhibition related targets using affinity methods of selection.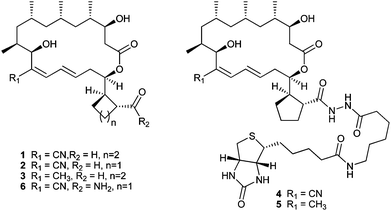 | ||
| Fig. 1 Structures of borrelidin, analogs and chemical probes. | ||
The resulting probes were used as bait for the biopanning of a T7Select cDNA phage display library derived from a human colon cancer cell line (Novagen) expressed in Escherichia coliBLT5403. This method has been used successfully with other natural products.11,12 After four rounds of biopanning with 4, the amplified lysate was split and two fifth round biopanning experiments were performed in parallel, one using SDS elution (as used for rounds 1–4) and the other using elution by E. colicells which has been suggested to increase the likelihood of identifying weak target![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :ligand binding interactions.12 This procedure resulted in the progressive enrichment of two phage products of approximately 450 and 600 base pairs as shown by PCR amplification of the complete phage pool eluted after each round of biopanning (Fig. 2a). For each fifth round elution experiment DNA from 12 individual phage plaques was amplified using PCR (Fig. 2b), sequenced and compared with published databases to identify the displayed protein fragments. Two out of the SDS-eluted phage contained a cDNA insert encoding a sequence encoding 99 amino acids in frame with the phage coat protein, of which a region of 83 residues was identical with human formin binding protein 21 (FBP21) (Fig. 2c). Strikingly, this protein fragment corresponds exactly to the tandem WW domains of FBP21, a structural motif likely to mediate specific protein-protein interactions.13 Of the remaining 10 SDS-eluted phage, none found a significant match in sequence databases. Most displayed 20 amino acids or less, and were disregarded as background binders, while the others were eliminated as they contained open reading frames reversed with respect to the direction of expression, or predicted a protein not in-frame with the phage coat protein. Of the E. coli-eluted phage, 10 contained the same FBP21 insert (see Fig. 2b). The remaining clones contained a cDNA insert of less than 150 bp in length that generated no BLAST hits. As a control, identical experiments were performed using 5 as bait. This gave a very different amplification pattern, and none of the phage contained FBP21 encoding inserts. When repeated, all experiments gave essentially the same results.
:ligand binding interactions.12 This procedure resulted in the progressive enrichment of two phage products of approximately 450 and 600 base pairs as shown by PCR amplification of the complete phage pool eluted after each round of biopanning (Fig. 2a). For each fifth round elution experiment DNA from 12 individual phage plaques was amplified using PCR (Fig. 2b), sequenced and compared with published databases to identify the displayed protein fragments. Two out of the SDS-eluted phage contained a cDNA insert encoding a sequence encoding 99 amino acids in frame with the phage coat protein, of which a region of 83 residues was identical with human formin binding protein 21 (FBP21) (Fig. 2c). Strikingly, this protein fragment corresponds exactly to the tandem WW domains of FBP21, a structural motif likely to mediate specific protein-protein interactions.13 Of the remaining 10 SDS-eluted phage, none found a significant match in sequence databases. Most displayed 20 amino acids or less, and were disregarded as background binders, while the others were eliminated as they contained open reading frames reversed with respect to the direction of expression, or predicted a protein not in-frame with the phage coat protein. Of the E. coli-eluted phage, 10 contained the same FBP21 insert (see Fig. 2b). The remaining clones contained a cDNA insert of less than 150 bp in length that generated no BLAST hits. As a control, identical experiments were performed using 5 as bait. This gave a very different amplification pattern, and none of the phage contained FBP21 encoding inserts. When repeated, all experiments gave essentially the same results.
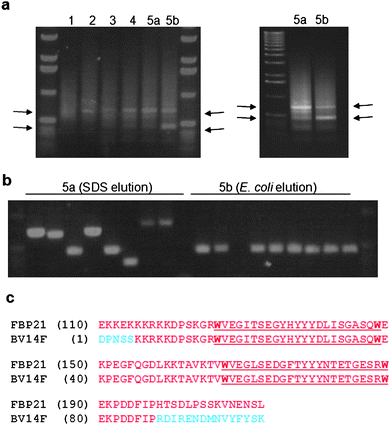 | ||
| Fig. 2 (a) Four serial rounds of biopanning resulted in the progressive enrichment of two phage products of approx. 450 and 600 bp respectively (highlighted by arrows) as judged by PCR of total phage after each round. The final round was eluted using either SDS (5a) or E. coli (5b), and resulted in differential enrichment of these two bands (see right hand image for clarity); (b) PCR analysis of single colony isolation of phage from the final round of biopanning (representatives of rounds 5a & b are shown); (c) Sequence analysis of the phage PCR products identified a common insert containing a translated 83 amino acid section (red) of FBP21 including its tandem WW domains (underlined); the translated sequence for a single clone BV14F is shown alongside that for FBP21 (NM_007187). | ||
FBP21 is a known splicing factor.13 The WW domains of FBP21 are similar to those of yeast splicing factor PRP40, and FBP21 is present in highly purified spliceosomal complex A. It co-localizes with splicing factors in nuclear speckles, and interacts directly with U1 snRNP protein U1C, the core snRNP splicing factors SmB and SmB′, and the branch point binding-protein SF1/mBBP, strongly suggesting that FBP21 plays a role in the cross-intron bridging of U1 and U2 snRNPs in the mammalian A complex. Recent studies have shown that FBP21 also interacts with SIPP1 (splicing factor that interacts with PQBP1 and PP1) in vitro and in vivo, and targeted mutagenesis has confirmed that these interactions are mediated by the tandem WW domains.14 Formin binding proteins (FBPs) and other WW domain-encoding proteins are increasingly viewed as scaffolding proteins which provide a platform for the exchange of spliceosome-associated splicing factors and other proteins.15,16 For example, the WW domain-containing proteins CA150 and FBP11 are known to associate with U2 snRNP, splicing factors SF1, U2AF and components of the SF3 complex,15 indicating that WW domain-associating factors bind to the 3′ part of an intron during spliceosome assembly. It was additionally shown that such effects can modulate alternative exon selection by facilitating particular 3′ splice site recognition, an effect apparently specific to selection of the 3′ site. Identification of the WW domains of FBP21 as a potential molecular target of 1 raised the possibility that inhibition of pre-mRNA splicing, and/or the modulation of alternative splicing, might account for at least part of the anti-angiogenic effect of this compound. Natural products have repeatedly shown their value as selective inhibitors/modulators/identifiers of cellular processes,17 and several natural products of polyketide origin have recently been reported as inhibitors of the spliceosome.18
To measure directly the binding of 1 and 2, a fragment of FBP21 (amino acids 129–197 of NP_061235.2) housing the tandem WW domains was expressed and purified as a GST (glutathione S-transferase) fusion protein as described previously.19 This fragment has been shown competent in binding various splicing factors, and utilized in a range of assays.13 The affinity of 1 and 2 was measured using isothermal titration calorimetry (ITC). Binding of both compounds was associated with only a small change in enthalpy, and was driven by favourable entropic effects (see ESI†). This entropic binding is consistent with a hydrophobic binding interaction at the ligand–protein interface, whereby the binding is presumably driven by desolvation of 1 and 2. Because the enthalpy changes associated with these interactions were small, optimal signal to noise ratios were obtained for experiments conducted at 15 °C. 1 and 2 gave dissociation constants (Kd) under these conditions of 11.8 ± 2.7 μM and 9.7 ± 3.1 μM respectively. The affinity of the control peptide SMB20 was 16.4 ± 1.9 μM under these conditions, and in contrast is entirely driven by enthalpy.
To explore the effect of 1 and analogs on alternative pre-mRNA splicing we focused on VEGF, a key regulator of physiological and pathological angiogenesis, which is organized as eight exons and exists in multiple isoforms generated by alternative pre-mRNA splicing.21 A C-terminal splicing event in the VEGF mRNA results in production of two families of VEGF isoforms, termed VEGFxxx and VEGFxxxb, which are formed by use of alternative 3′ acceptor splice sites in exon 8, 66-bp apart. In both families of isoforms, alternative splicing results in proteins with identical numbers of amino acid residues but the mRNAs differ in an 18 base open reading frame encoding for either the pro-angiogenic (exon 8a) Cys-Asp-Lys-Pro-Arg-Arg or the anti-angiogenic (exon 8b) Ser-Leu-Thr-Arg-Lys-Asp at the C-terminus.22,23 In contrast to the VEGFxxx family, which is potently pro-angiogenic, the VEGFxxxb family of proteins has anti-angiogenic activity and inhibits VEGF-mediated activation of VEGFR2 and tumour growthin vivo.23,24 VEGFxxxb isoforms are expressed in most tissues and by many cell types including retinal cells and can be differentially regulated in angiogenic conditions such as diabetic retinopathy25 and cancer.26 Thus, the expression ratio of these isoforms correlates strongly with the angiogenic status of cell types and is regulated differentially by splicing and growth factors.27
The effect of 1–3 and 6 on VEGF alternative splicing was examined in primary retinal pigmented endothelial (RPE) cells which express both VEGFxxx and VEGFxxxb isoforms (where xxx is used to denote the number of amino acids in the resulting protein, e.g.VEGF165, VEGF121b, or VEGF165b).27 RPE cells were treated with compounds and the culture medium analyzed after 72 h using an ELISA method as described previously.27 At concentrations of 0.5 and 5 μM 1 and 2 significantly decreased the expression of VEGFxxx isoforms whilst increasing the expression of VEGFxxxb isoforms (Fig. 3a, b). The VEGFxxxb isoform predominantly expressed was VEGF165b although VEGF121b was also seen at a lower level. The inactive compound 3 had no effect, even at 5 μM. Upon treatment with increasing concentrations of 2 RPE cells showed a concentration-dependent decrease in expression of VEGFxxx (calculated from the difference between total VEGF and VEGFxxxb) and a concomitant increase in VEGFxxxb (Fig. 3c) (2 was used in preference to 1 here and in further studies described due to its lower toxicity to RPE cells). VEGF165b competitively antagonises VEGF165-mediated endothelial proliferation, migration and angiogenesis at or above a 1:1 molar ratio.29 A value >1 represents a pro-angiogenic cellular environment, whilst a value <1 represents an anti-angiogenic environment. A VEGF165:VEGF165b molar ratio of 5 should be sufficient to support angiogenesis, and a value of 0.2 sufficient to block angiogenesis completely.23 As shown in Fig. 3c treatment with 2 at ≥0.5 μM shifted this ratio to a clearly anti-angiogenic value (<1). It is noteworthy that the amide analog 6 proved to be as effective as 1 and 2 in stimulating VEGFxxxb protein expression, confirming that modification of the carboxylic acid group, as in the biotinylated probes 4 and 5, does not compromise the activity of the compound in modulating splicing.
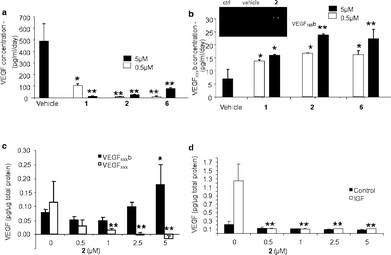 | ||
| Fig. 3 Comparison of the effect of 0.5 and 5 μM 1 and 2 on total VEGF (a) and VEGFxxxb (b) concentrations in RPE cells measured by ELISA, *p < 0.05, **p < 0.01, Dunnett's compared with vehicle, n = 3; the insert shows PCR for VEGF165b in control cells, vehicle treated and cells treated with 228(c) RPE cells treated with increasing concentrations of 2 for 24 h; VEGFxxxb and total VEGF concentrations measured by ELISA, *p < 0.05, Dunnett's, **p < 0.01, n = 3; (d) Effect of IGF-1 and 2 treatment on VEGFxxxprotein levels; RPE cells were treated with increasing concentrations of 2 and 1μM IGF-1 for 24 h; total VEGF levels measured by ELISA, p = 0.0113, one way ANOVA, **p < 0.01, Dunnett's compared with IGF alone, n = 3. | ||
Treatment of RPE cells with insulin-like growth factor-1 (IGF-1) (1 μM) upregulates VEGF165, by inducing proximal splice site selection.27 When 2 was co-administered at several concentrations with IGF-1 it completely abolished this effect upon VEGF165 protein expression (Fig. 3d), showing that proximal splice site selection in the VEGF gene induced by IGF-1 was being inhibited. The lack of upregulation of any VEGF isoform by IGF-1 in the presence of 2 indicates that IGF was prevented from stimulating selection of the proximal splice site. To further probe the interaction between 2 and FBP21, we transfected RPE cells with the FBP21 cDNA expression plasmid pWV327, a pCMV6 based vector for expression of full-length FBP21 (see ESI†). RPE cells transfected with pWV327 produced significantly less VEGF165b (0.36 ± 0.03 pg/μg protein) than cells transfected with the control vector (0.49 ± 0.03 pg/μg protein, p < 0.05; Fig. 4a). The upregulation of VEGF165b by 2 seen in control cells (2.9 ± 0.78 fold) was reduced by transfection with FBP21 (1.8 ± 0.15 fold, p = 0.05), indicating that the response to 2 is likely to be FBP21-dependent (Fig. 4b). An FBP21-dependent response to 2 is consistent with the concept that it acts through direct binding to FBP21, although a more indirect effect cannot be completely excluded.
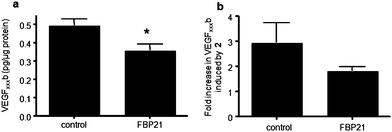 | ||
| Fig. 4 Enhanced FBP21 expression mitigates the effect of 2 on alternative splicing of VEGF in RPE cells. RPE cells were transfected with either control vector or FBP21 expression vector. (a) ELISA for VEGFxxxb using biotinylated VEGFxxxb specific detection antibody, *p < 0.05 compared with control, Paired t test, n = 3; (b) Transfected cells were then treated with 0.5μM 2 for 72 h and protein assayed by ELISA; results are expressed relative to vehicle (0.1% DMSO) treated cells, p = 0.05 Mann Whitney U test, n = 4. | ||
Conclusions
The modulation of VEGF alternative splicing in RPE cells by 2 was both reproducible, statistically significant, and dose dependent. Moreover, analog 3, which is neither cytotoxic nor an inhibitor of angiogenesisin vitro, was not active in this assay. It is important to note, however, that the ability of 1 and 2 to modulate VEGF splicing requires μM concentrations which are significantly higher than those required for anti-angiogenic activity measured previously by monitoring capillary and tubule formation in vitro (nM for 1 and pM for 2).3,5 It has been proposed that 1 operates through (at least) two distinct pathways to exert its anti-angiogenic activity, one of which appears dependent upon its known ability to inhibit, or bind to, ThrRS.9 We now propose that another pathway may be through spliceosome inhibition and/or modulation.In the course of our studies we identified the splicing factor FBP21 as a potential molecular target of 1 through the use of affinity biopanning experiments, and confirmed that 1 and 2 bind to its tandem WW domains at concentrations required for modulation of VEGF alternative splicing in RPE cells. It was additionally shown that expression of FBP21 in RPE cells was capable of modulating terminal exon alternative splicing, significantly lowering the levels of VEGF165b, while the upregulation of VEGF165b induced by treating RPE cells with 2 was abrogated by the expression of FBP21. Additionally, 2 potently inhibited the ability of IGF-1 to upregulate VEGF165 expression through induction of proximal splice site selection.
While still preliminary, these data are consistent with a model in which FBP21 is a splicing factor, one function of which is the selection of specific terminal (exon-8) splice sites during VEGF alternative splicing, and that 1 and 2 can modulate this process through interaction with FBP21. The regulation of VEGF splicing is not well understood. Currently the only splicing factors to be implicated in terminal exon splicing of VEGF are SRp55, (which promotes distal splicing), ASF/SF2 and SRp40 (which promote proximal splicing).27 Further work will be required to confirm the role of the 1-FBP21 interaction both for VEGF and more generally at influencing the outcome of alternative splicing. It is an attractive hypothesis that concerted regulation of the switch to a pro-angiogenic state might involve alteration in splicing patterns of multiple target proteins. Great strides have been made recently in reconstituting spliceosomal activities in vitro,30,31 and use of such in vitro assays should in future allow a precise evaluation of the molecular mechanisms involved in 1 action. Conversely, the use of 1 derived compounds offers the perspective of deconvoluting individual steps in the action of the spliceosome.30
The findings reported here demonstrate once again the tremendous value of natural products both as chemical tools for uncovering new and rich biology, and as lead compounds for the development of potential therapeutic agents.17 Indeed, we suggest that interfering with interactions between FBP21 and its normal binding partners represents a novel pharmacological target. It may be possible in the future to obtain 1-based compounds that are selective for one target over the other, e.g.FBP21 over ThrRS. We believe that detailed evaluation of 2 and other analogs is warranted as a source of potential therapeutic intervention for vascular diseases.
Experimental procedures
General chemical methods, natural products chemistry & chemical synthesis
These are detailed in the ESI†.In vitro HUVEC anti-angiogenesis assays
Angiogenesis assays with 1 and 3 were carried out at the angiogenesis resource center of the NCI and details of the assay protocols and results are given in the ESI†.Isothermal titration calorimetry
Purified GST-FBP fusion protein19,20 was exhaustively dialysed overnight at 4 °C against 100 mM phosphate saline buffer (pH 7.2 NaCl 140 mM KCl 2.7 mM). Stock solutions of SMB, 1 and 2, were lyophilised/evaporated and re-dissolved in the exact same dialysis buffer. Samples were degassed and ITC was performed at 15 °C using a VP-ITC microcalorimeter (Microcal, Inc). SMB, 1 and 2 were titrated into the sample cell containing GST-FBP in 20 injections of 15 μL with reference power of 2 kcal s−1. Data analysis was performed using Origin 5.0 (Origin Laboratories) by the single-site binding model. The fitted data allows Kd and ΔH to be determined directly, which, using the relationship ΔG = -RT ln Ka = ΔH - T ΔS, allows the entropy change (TΔS) and the free energy of binding (ΔG) to be calculated. The data obtained are presented in the ESI†.Biopanning of cDNA phage display library
Streptavidin wells were incubated with 1 μM of probe at 4 °C overnight to prepare the wells for biopanning. After incubation, unbound biotinylated probe was removed from the well by washing with phosphate buffered saline (PBS). Wells were then blocked with blocking solution (3% skimmed milk in PBS) for at least one hour at room temperature. Blocked wells were then briefly washed with PBS and were ready for biopanning use. The Novagen T7 colon cancer cell line cDNA phage library was amplified using standard techniques (see Novagen T7Select™ manual provided with Novagen phage library) and stored either in 0.5 M NaCl at 4 °C (for short term storage) or in 8% glycerol at −80 °C (for long term storage).Amplified phage libraries were titered using standard techniques and were found to contain an average of 5 × 1010 plaque forming units per mL (pfu mL−1). In the first round of biopanning, amplified phage library (0.15 mL) was added to an empty streptavidin coated well and incubated at room temperature for at least 1 h. This step was included to deselect any phage displaying streptavidin binding proteins. After incubation, the phage were removed from the well and mixed with 4x blocking solution (12% skimmed milk in PBS) (50 μL) and incubated at room temperature for at least 1 h.
Blocked phage library (125 μL) was added to each pre-prepared biotinylated well and incubated at room temperature for at least 1 h. The supplied library contains an estimated 1.7 x107 distinct primary recombinant phage clones so >200 copies of each clone were added to the well. After incubation unbound phage were removed from the well with wash solution. The wash solution was varied depending on the round of biopanning. For the first round of biopanning PBS1 was used. For subsequent rounds the stringency of the wash was increased by using sequentially PBS2, PBS3 and PBS4: PBS1 (round 1), 150 mM NaCl, 0.1% Tween20 in PBS; PBS2 (round 2), 170 mM NaCl, 0.3% Tween20 in PBS; PBS3 (round 3), 200 mM NaCl, 0.5% Tween20 in PBS; PBS4 (round 4), 250 mM NaCl, 0.7% Tween20 in PBS. During the first biopanning round, ten washes were performed with PBS1 followed by five washes with PBS to remove all traces of Tween20 and NaCl from the well. Similarly for subsequent rounds ten washes were performed with the appropriate wash solution followed by 5 washes with PBS. After rounds 1–4 the wells were eluted with SDS elution solution: 1% SDS in PBS (100 μL) was added to the well and incubated at room temperature for at least 1 h. The eluent was then removed from the well and pipetted into E. coliBLT5403 grown in 2xTY media to an OD = 0.4 (50 mL in a 250 mL conical flask). The flask was incubated at 37 °C with shaking at 250 rpm for approximately 4 h or until complete lysis had occurred. The lysate was transferred to a 50 mL falcon tube, solid NaCl was added to a final concentration of 0.5 M, and the lysate was centrifuged at 4000 rpm for 10 min. The supernatant was transferred to a new 50 mL tube and was stored at 4 °C or used in a subsequent round of biopanning.
Two different elution techniques were employed to remove bound phage from the wells in round 5. (a) SDS elution: SDS elution solution (1% SDS in PBS) (100 μL) was added to the well and incubated at room temperature for at least 1 h; or (b) E. coli elution:12E. coli elution solution (the hostE. coli strain (BLT5403) grown to OD = 0.4) (100 μL) was added to the well and incubated at room temperature for at least 1 h. In each case the eluant was treated as described above.
Lysates from the separate round 5 panning experiments were serially diluted, mixed with E. coli culture (BLT5403 grown overnight at 37 °C) (250 μL), mixed with melted top agarose (3 mL, pre-equilibrated to 45 °C) and poured onto a pre-warmed plate of 2TY agar. Plates were incubated at 37 °C for 2–4 h until a bacterial lawn containing phage plaques was seen. Twelve individual phage plaques from each round 5 experiment were cut from the plates and placed into tubes containing PBS (200 μL). Tubes were vortexed briefly to liberate the phage from the agar plug. Each phage plaque suspension (1 μL) was then used as the template in a PCR reaction using T7SelectUP: 5′-GGAGCTGTCGTATTCCAGTC and T7SelectDOWN: 5-AACCCCTCAAGACCCGTTTA primers, Taq DNA polymerase and the following PCR program: 1 cycle: 80 °C for 10 min; 30–35 cycles: 94 °C for 1 min, 55 °C for 1 min, 72 °C for 2 min. The resulting PCR products were cleaned and sequenced with T7SelectUP and T7SelectDOWN oligos and the resultant data were submitted for nucleotide BLASTN searches on the NCBI website. Predicted displayed amino acid sequences were submitted to BLASTP.
RPE cell culture and VEGF experiments
These are detailed in the ESI†.Acknowledgements
JW is funded by the Wellcome Trust (079663/Z/06/Z) and DOB by the British Heart Foundation (BS/06/005). This work was funded in part by a grant from the Richard Bright VEGF Research Trust. Mark Bedford is supported by a Welch Foundation Grant (G-1495) and a NIDA grant (DA025800). Astrid Spannhoff is supported by The German Research Foundation (SP1262/1-1).Notes and references
- S. J. Moss, I. Carletti, C. Olano, R. M. Sheridan, M. Ward, V. Math, M. Nur-e-Alam, A. F. Braña, M.-Q. Zhang, P. F. Leadlay, C. Méndez, J. A. Salas and B. Wilkinson, Chem. Commun., 2006, 2341–2343 RSC.
- M. Buck, A. C. Farr and R. J. Schnitzer, Trans. New York Acad. Sci., 1949, 11, 207–210 Search PubMed.
- T. Wakabayashi, R. Kagayama, N. Naruse, N. Tsukahara, Y. Funahashi, K. Kitoh and Y. Watanabe, J. Antibiot., 1997, 50, 671–676 CAS.
- Y. Funahashi, T. Wakabayashi, T. Semba, J. Sonoda, K. Kitoh and K. Yoshimatsu, Oncol. Res., 1999, 11, 319–329 CAS.
- B. Wilkinson, M. A. Gregory, S. J. Moss, I. Carletti, R. M. Sheridan, A. Kaja, M. Ward, C. Olano, C. Méndez, J. A. Salas, P. F. Leadlay, R. VanGinkel and M.-Q. Zhang, Bioorg. Med. Chem. Lett., 2006, 16, 5814–5817 CrossRef CAS also see ESI.
- N. Makk, G. Ambrus, A. Tegdes, A. Jeney and F. Timar, PCT., WO 01/09112, 2001.
- R. Harisi, I. Kenessey, J. N. Olah, F. Timar, I. Babo, G. Pogany, S. Paku and A. Jeney, Anticancer Res., 2009, 29, 2981–2985.
- B. Ruan, M. L. Bovee, M. Sacher, C. Stathopoulos, K. Poralla, C. S. Francklyn and D. Söll, J. Biol. Chem., 2005, 280, 571–577 CAS.
- T. Kawamura, D. Liu, M. J. Towle, R. Kageyama, N. Tsukahara, T. Wakabayashi and B. A. Littlefield, J. Antibiot., 2003, 56, 709–715 CAS.
- S. C. Gerken and S. M. Arfin, J. Biol. Chem., 1984, 259, 9202–9206 CAS.
- H. Kim, L. Deng, X. Xiong, W. D. Hunter, M. C. Long and M. C. Pirrung, J. Med. Chem., 2007, 50, 3423–3426 CrossRef CAS; Y. Jin, J. Yu and Y.-G. Yu, Chem. Biol., 2002, 9, 157–162 CrossRef CAS.
- K. M. McKenzie, E. J. Videlock, U. Splittgerber and D. J. Austin, Angew. Chem. Intl. Ed. Engl., 2004, 43, 4052–4055 CrossRef CAS.
- M. T. Bedford, R. Reed and P. Leder, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 10602–10607 CrossRef CAS.
- X. Huang, M. Beullens, J. Zhang, Y. Zhou, E. Nicolauescu, B. Lesage, Q. Hu, J. Wu, M. Bollen and Y. Shi, J. Biol. Chem., 2009, 284, 25375–25387 CrossRef CAS.
- K.-T. Lin, R.-M. Lu and W.-Y. Tarn, Mol. Cell. Biol., 2004, 24, 9176–9185 CrossRef CAS.
- R. J. Ingham, K. Colwill, C. Howard, S. Dettwiler, C. S. M. Lim, J. Yu, K. Hersi, J. Raaijmakers, G. Gish, G. Mbamalu, L. Taylor, B. Yeung, G. Vassilovski, M. Amin, F. Chen, L. Matskova, G. Winberg, I. Ernberg, R. Linding, P. O'Donnell, A. Starostine, W. Keller, P. Metalnikov, C. Stark and T. Pawson, Mol. Cell. Biol., 2005, 25, 7092–7106 CrossRef CAS.
- N. Dixon, L. S. Wong, T. H. Geerlings and J. Micklefield, Nat. Prod. Rep., 2007, 24, 1288–1310 RSC.
- Y. Kotake, K. Sagane, T. Owa, Y. Minori-Kiyosue, H. Shimizu, M. Uesugi, Y. Ishihama, M. Iwata and Y. Mizui, Nat. Chem. Biol., 2007, 3, 570–575 CrossRef CAS; D. Kaida, H. Motoyoshi, E. Tashiro, T. Nojima, M. Hagiwara, K. Ishigami, H. Watanabe, T. Kitahara, T. Yoshida, H. Nakajima, T. Tani, S. Horinouchi and M. Yoshida, Nat. Chem. Biol., 2007, 3, 576–583 CrossRef CAS; K. O'Brien, A. J. Matlin, A. M. Lowell and M. J. Moore, J. Biol. Chem., 2008, 283, 33147–33154 CrossRef CAS.
- D. C. Chan, M. T. Bedford and P. Leder, EMBO J., 1996, 15, 1045–1054 CAS.
- M. T. Bedford, D. C. Chan and P. Leder, EMBO J., 1997, 16, 2376–2383 CrossRef CAS; A. Espejo, J. Cote, A. Bednarek, S. Richard and M. T. Bedford, Biochem. J., 2002, 367, 697–702 CrossRef CAS.
- S. J. Harper and D. O. Bates, Nat. Rev. Cancer, 2008, 8, 880–887 CrossRef CAS.
- D. O. Bates, T. G. Cui, J. M. Doughty, M. Winkler, M. Sugiono, J. D. Shields, D. Peat, D. Gillat and S. J. Harper, Cancer Res., 2002, 62, 4123–4131 CAS.
- J. Woolard, W.-J. Wang, H. S. Bevan, Y. Qiu, L. Morbidelli, R. O. Pritchard-Jones, T.-G. Cui, M. Sugiono, R. E. Waine, R. Perrin, R. Foster, J. Digby-Bell, J. D. Shields, C. E. Whittles, R. E. Mushens, D. A. Gillatt, M. Ziche, S. J. Harper and D. O. Bates, Cancer Res., 2004, 64, 7822–7835 CrossRef CAS.
- S. Cébe Suarez, M. Pieren, L. Cariolato, S. Arn, U. Hoffmann, A. Bogucki, C. Manlius, J. Wood and K. Malner-Hofer, Cell. Mol. Life Sci., 2006, 63, 2067–2077 CrossRef CAS.
- R. M. Perrin, O. Konopatskya, Y. Qiu, S. Harper, D. O. Bates and A. J. Churchill, Diabetologia, 2005, 48, 2422–2427 CrossRef CAS.
- D. O. Bates, D. P. MacMillan, J. G. Manjaly, Y. Qiu, S. J. Hudson, H. S. Bevan, A. J. Hunter, P. W. Soothill, M. Read, L. F. Donaldson and S. J. Harper, Clin. Sci. (London), 2006, 110, 575–585 Search PubMed; R. O. Pritchard-Jones, B. D. Dunn, Y. Qiu, A. H. Varey, A. Orlando, H. Rigby, S. J. Harper and D. O. Bates, Br. J. Cancer, 2007, 97, 223–230 CrossRef CAS.
- D. G. Nowak, J. Woolard, E. M. Amin, O. Konopatskya, M. A. Saleem, A. J. Churchill, M. R. Ladomery, S. J. Harper and S. J. Harper, J. Cell Sci., 2008, 121, 3487–3495 CrossRef CAS.
- Y. Qui, C. Hoareau-Aveilla, S. Oltean, S. J. Harper and D. O. Bates, Biochem. Soc. Trans., 2009, 37, 1207–1213 CrossRef.
- E. S. Rennel, E. Waine, H. Guan, Y. Schüler, W. Leenders, J. Woolard, M. Sugiono, D. Gillatt, E. S. Kleinerman, D. O. Bates and S. J. Harper, Br. J. Cancer, 2008, 98, 1250–1257 CrossRef CAS.
- M. S. Jurica, Nat. Chem. Biol., 2008, 4, 3–6 CrossRef CAS.
- N. Kataoka and G. Dreyfuss, Methods Mol. Biol., 2008, 488, 357–365 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: General chemical methods; natural products & synthetic chemistry; isothermal titration calorimetry data; in vitro HUVECanti-angiogenesis assays; RPE cell culture and VEGF experiments; correction of the FBP21 cDNA expression vector. See DOI: 10.1039/c0sc00297f |
| This journal is © The Royal Society of Chemistry 2011 |