Rhodium catalyzed allene amidation: a facile entry into 2-amidoallylcations for unusual [3 + 3] annulation reactions†
Armin H.
Stoll
and
Simon B.
Blakey
*
Department of Chemistry, Emory University, Atlanta, GA, U.S.A. E-mail: sblakey@emory.edu
First published on 16th September 2010
Abstract
The interaction of a sulfamate ester derived metallonitrene with an allene generates a versatile intermediate with 2-amidoallylcation like reactivity. In this article we outline reactivity patterns for this novel dipolar species, demonstrating both [3 + 2] reactions with benzaldehyde, and unusual [3 + 3] annulation reactions with a variety of nitrones.
Introduction
In the arena of complex molecule synthesis, the most useful transformations are those that lead to substantial increases in molecular complexity.1 In this context, cycloaddition reactions have long been recognized as particularly powerful. Their ability to construct multiple bonds in a predictable, stereodefined manner has ensured that they have been employed at the crux of many important syntheses.2 Despite the depth of knowledge regarding the Diels–Alder transformation as well as 1,3-dipolar cycloadditions existing within the community,3,4 the development of new cycloaddition processes to access novel molecular frameworks remains a vibrant area of contemporary research.5Amongst these recent advances, the development of nitrones as suitable coupling partners in homo [3 + 2] reactions stands out as a powerful method for the construction of stereodefined heterocycles.6,7
As a part of our program investigating cascade reactions initiated by electrophilic metallonitrenes, we recently reported a new method for the diastereoselective synthesis of highly substituted aminocyclopropanes and postulated that the reactions proceeded via 2-amidoallylcation intermediates (Scheme 1).8
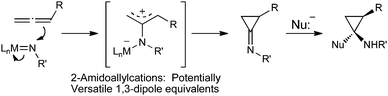 | ||
| Scheme 1 Allene amidation with electrophilic metallonitrenes: a general approach to 2-amidoallylcations. | ||
Consistent with this hypothesis, we observed that intramolecular oxidative sulfamidation of allene 1 in the presence of five equivalents of benzaldehyde (2) resulted in the formation of trisubstituted 3-aminotetrahydrofuran 3 (Scheme 2). We propose that this product is formed in a [3 + 2] cycloaddition between the aldehyde and the intermediate 2-amidoallylcation.
![3-Aminotetrahydrofuran synthesis viaallene amidation/[3 + 2] cycloaddition cascade.](/image/article/2011/SC/c0sc00375a/c0sc00375a-s2.gif) | ||
| Scheme 2 3-Aminotetrahydrofuran synthesis viaallene amidation/[3 + 2] cycloaddition cascade. | ||
These 2-amidoallylcations resemble trimethylenemethane (TMM) and its synthetic equivalents, powerful synthons for the stereoselective construction of five-membered rings,9 as well as the related 2-oxyallylcation species (e.g.2-alkoxy-, 2-metallo-oxyallyl- or 2-silyloxyallylcations).10 In contrast to the well-established methods for accessing these reactive intermediates, the preparation of 2-amidoallylcations has received far less attention. Although both α-chloroimine and α-chloroenamine activation through halide abstraction with stochiometric silver(I) salts have been investigated, the instability of the starting materials prevented widespread use of this methodology.11 Recently, the Shipman group discovered that Lewis acid induced ring opening of strained methyleneaziridines provided 2-amidoallylcation like intermediates that, in an analogous fashion to their 2-oxyallylcation counterparts, were capable of engaging tethered dienes in [4 + 3] cycloaddition reactions.12 In spite of these developments, truly general methods for the preparation of 2-amidoallylcation intermediates and the subsequent exploration of 2-amidoallylcation reactivity remain elusive.
In this report we present the results of our study into the in situ generation of 2-amidoallylcations viaallene sulfamidation in the context of novel [3 + 3] annulation reactions with nitrones as complimentary 1,3-dipolar reagents.
Results and discussion
Our investigations into cascade reactions initiated by metallonitrene interactions with π-systems have demonstrated that the sulfamate esters developed by Du Bois for C–H amination reactions were ideally suited for this chemistry and preferentially cyclized to provide seven-membered rings.8,13,14 In light of these results, we adopted homoallenylsulfamate ester 4 for our exploratory work (Scheme 3). Oxidation of 4 with PhI(O2CtBu)2 in the presence of catalytic Rh2(esp)2 and four equivalents of N,α-diphenyl nitrone 5 gave rise to the unexpected bridged bicyclic heterocycle 6 in 65% yield. In this reaction the nitrone had added across the nitrogen and the most substituted carbon of the presumptive 2-amidoallylcation intermediate. We did not detect any of the expected [3 + 3] addition product 7 that would arise from the addition of the nitrone across the two carbon termini of the 2-amidoallylcation.![In situ generation of a 2-amidoallylcation facilitating a regio- and diastereoselective [3 + 3] annulation reaction.](/image/article/2011/SC/c0sc00375a/c0sc00375a-s3.gif) | ||
| Scheme 3 In situ generation of a 2-amidoallylcation facilitating a regio- and diastereoselective [3 + 3] annulation reaction. | ||
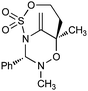
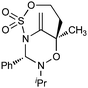
Despite their significant structural differences, regioisomers 6 and 7 were difficult to distinguish by 1H and 13C NMR alone. In order to confirm our structural assignment, adduct 6 was subjected to standard hydroboration/oxidation conditions to provide crystalline derivative 8 (Scheme 4). The structure of 8 was determined unambiguously by X-ray crystallography.
![Hydroboration of [3 + 3] annulation product 6 allows unambiguous structural assignment by X-ray crystallography.](/image/article/2011/SC/c0sc00375a/c0sc00375a-s4.gif) | ||
| Scheme 4 Hydroboration of [3 + 3] annulation product 6 allows unambiguous structural assignment by X-ray crystallography. | ||
A brief survey of reaction conditions in which alternative dirhodium(II) tetracarboxylate salts (Rh2(OAc)4, Rh2(OPiv)4 and Rh2(O2CCPh3)4), and solvents (C6H6, CH2Cl2) were tested did not provide any improvement in the conversion of allene 4 to the bicyclic annulation product 6. Furthermore, the regioselectivity remained constant as the reaction parameters were varied, and in all cases 6 was the only detectable addition product in the reaction mixtures.
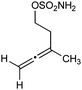
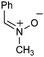
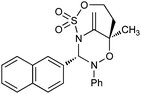
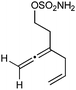
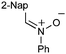
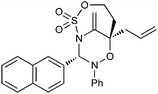
Having established that the unusual regiochemical outcome of this [3 + 3] addition reaction was not impacted by the nature of the dirhodium(II) catalyst, we proceeded to investigate the scope of the reaction with respect to both nitrone and allene structure (Table 1). Our studies revealed that a range of 1,1-disubstituted allenes with moderately sized substituents (methyl (entries 1–4), allyl (entries 5–7) and benzyl (entry 8)) were compatible with our protocol. In all cases the products were isolated as single regio- and diastereomers.
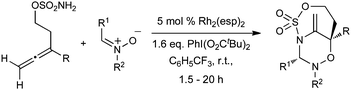
| Entry | Allene | Nitrone | Product | Yield |
|---|---|---|---|---|
| 1 |
|
|
|
68% |
| 2 |

|
|
|
32% |
| 3 |
|
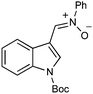
|
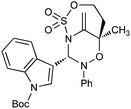
|
46% |
| 4 |
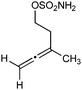
|
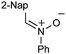
|
|
62% |
| 5 |
|
|
|
60% |
| 6 |
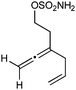
|
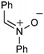
|

|
69% |
| 7 |
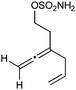
|
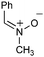
|
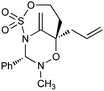
|
73% |
| 8 |
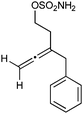
|
|

|
48% |
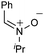
With respect to the nitrones, aryl- and heteroaryl aldehyde derived nitrones (phenyl- (entries 1, 2, 6–8), 2-napthyl- (entries 4,5) and 3-N-Boc-indoyl- (entry 3)) are generally well tolerated in the [3 + 3] annulation reactions. However nitrones prepared from alkyl-substituted aldehydes are not suitable dipolarophiles for the 2-amidoallylcations generated under these reaction conditions (not shown). Products derived from nitrones bearing aryl, or small alkyl substituents on the nitrogen atom could be accessed in good yields (46–73%). However larger alkyl substituents (e.g.iPr) resulted in reduced addition efficiencies (entry 2, 32%).
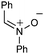
In the extreme case, no addition product was observed when the very bulky N-tBu-nitrone 9 was evaluated (Scheme 5). Instead, the 2-amidoallylcation collapsed to form an N-sulfonylimino-cyclopropane that was subsequently trapped by the pivaloate released from the hypervalent iodine oxidant to give N-sulfamoyl-O-pivaloylcyclopropylaminol 10 in 60% yield (93![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :7 d.r.).15
:7 d.r.).15
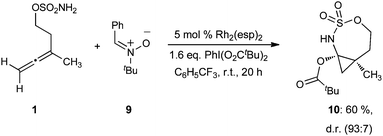 | ||
| Scheme 5 Oxidative rearrangement of allene sulfamate ester and trapping with pivaloate in the presence of bulky N-tBu-nitrone. | ||
In addition to 1,1-disubstituted homoallenic sulfamate esters we also explored the reactivity of 1,1,3-trisubstituted allene 11. In this case a change in regioselectivity was observed with both N-aryl (5) and N-alkyl (12) nitrones, providing bicyclic products 13 and 14 respectively (Scheme 6). Consistent with our previous observations, the electrophilic carbon of the nitrone is bound to the nitrogen of the sulfamate ester in the product. However, in this case the nucleophilic oxygen of the nitrone interacts with the disubstituted terminus of the allene, suggesting that the regioselectivity of the reaction is governed by electronics, with the nitrone oxygen interacting with the allene carbon most capable of stabilizing a buildup of positive charge.
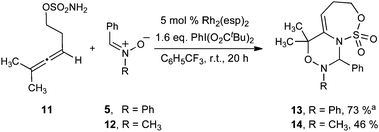 | ||
| Scheme 6
Allene amidation with electrophilic metallonitrenes: a general approach to 2-amidoallylcations. (a Isolated as a 9:1 mixture of regioisomers.) | ||
Furthermore, we have shown it is possible to link the sulfamoyl moiety to the allene through a nitrogen tether (15, 16),16 providing the alternative heterocycles 17 and 18 (Scheme 7). This alternative linker does not affect the regioselectivity observed in the annulation reaction.
![Amidation/[3 + 3] cycloaddition products from alternatively used 1,1-disubstituted allene sulfamides.](/image/article/2011/SC/c0sc00375a/c0sc00375a-s7.gif) | ||
| Scheme 7 Amidation/[3 + 3] cycloaddition products from alternatively used 1,1-disubstituted allene sulfamides. | ||
The annulation reactions described in this report consistently deliver products in which the nitrone has added across the nitrogen and a terminal carbon of the presumptive 2-amidoallylcation intermediate. This regioselectivity stands in contrast to that reported for [4 + 3] cycloadditions of 2-oxyallylcations, [4 + 3] cycloadditions of 2-amidoallylcations,12 and our own observations trapping the 2-amidoallylcation with benzaldehyde (Scheme 2), all of which provide products arising from addition across the two carbon termini of the reactive intermediate. These observations have caused us to carefully consider the mechanism of this reaction, and in particular the timing of the bond forming events (Scheme 8).17 Initial interaction of the metallonitrene (19) obtained by oxidation of the sulfamate ester in the presence of the dirhodium(II) tetracarboxylate catalyst is likely to produce a 2-amidoallylcation like intermediate (20). Although we cannot completely rule out the intermediacy of a methylene aziridine, neither we nor Robertson have observed such species when working with sulfamate esters.8,17a This stands in stark contrast to Robertson's observations with carbamate initiated allene aminations, which do produce observable methylene aziridine products.17b We suggest that intermediate 20 can interact with small reactive species such as benzaldehyde, leading to the observed [3 + 2] product in which the benzaldehyde has added across the two carbon termini of the intermediate (3).
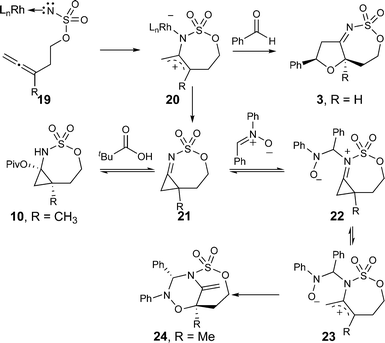 | ||
| Scheme 8 Proposed mechanism that accounts for the unusual regioselectivity observed in the allene amidation/nitrone annulation reaction. | ||
However, we speculate that the nitrones described in this study are not kinetically competent to react with this intermediate. Instead, the 2-amidoallylcation 20 rearranges to form a strained N-sulfonylimino-cyclopropane (21). In the absence of an external nitrone, or, if the nitrone is too sterically encumbered (e.g.9, Scheme 5), then the imine is trapped by the pivaloate biproduct from the hypervalent iodine oxidant, releasing the ring strain and leading to the observed aminol product 10.
However, in the presence of a nitrone, the strained cyclopropylimine (21) can attack the electrophilic carbon of the nitrone providing an electronic driving force (in addition to the strain release) facilitating cyclopropane ring opening (22 → 23).18 The oxygen of the nitrone then traps the resulting allylcation, interacting with the carbon most able to stabilize the carbocation.17
Consistent with this hypothesis, we note that when cyclopropylaminol 10 was reacted with benzaldehyde in the presence of scandium(III) triflate, the aldehyde was added across the nitrogen and carbon of the ring opened cyclopropane (25, Scheme 9). We believe that after pivaloate abstraction, the reaction follows an analogous pathway to that outlined for the nitrone annulation reactions, with imine attack on the aldehyde necessary to induce opening of the cyclopropane. We do not observe any of the [3 + 2] addition product 26 in which the aldehyde adds across the two carbon termini of an intermediate 2-amidoallylcation, analogous to the product obtained when an aldehyde is reacted directly with the 2-amidoallylcation formed by in situallene amidation.
![Cyclopropyl aminol ring opening provides a regioisomeric [3 + 2] annulation product.](/image/article/2011/SC/c0sc00375a/c0sc00375a-s9.gif) | ||
| Scheme 9 Cyclopropyl aminol ring opening provides a regioisomeric [3 + 2] annulation product. | ||
Conclusions
In conclusion, we have established that homoallenyl sulfamate esters readily undergo rhodium catalyzed oxidative allene amidation reactions, furnishing highly reactive 2-amidoallylcation intermediates. These 2-amidoallylcations are capable of participating in [3 + 2] addition reactions, but can also rearrange to form strained cyclopropyl imines. It is likely that the unusual regioselectivity in the [3 + 3] annulations with nitrones occurs as a result of nitrone interaction with the cyclopropyl imine, rather than a direct addition to the initially formed 2-amidoallylcation. The cascade allene amidation/nitrone addition reaction provides access to a variety of novel densely-functionalized heterocyclic scaffolds.Acknowledgements
Financial support was provided by the NSF (CHE 0847258). We thank Kenneth Hardcastle, Rui Cao and Sherry Lense for assistance with X-ray crystallography.Notes and references
- A. P. A. Wender, S. T. Handy and D. L. Wright, Chemistry & Industry, 1997, 765 Search PubMed.
- (a) C. P. Dell, J. Chem. Soc., Perkin Trans. 1, 1998, 1, 3873–3905 Search PubMed; (b) J. P. A. Harrity and O. Provoost, Org. Biomol. Chem., 2005, 3, 1349–1358 RSC; (c) Cycloaddition Reactions in Organic Synthesis (Eds: S. Kobayashi, K. A. Jorgensen) WILEY-VCH, Weinheim, 2001 Search PubMed.
- (a) K. C. Nicolaou, S. A. Snyder, T. Montagnon and G. Vassilikogiannakis, Angew. Chem., Int. Ed., 2002, 41, 1668–1698 CrossRef CAS; (b) G. Brieger and J. N. Bennett, Chem. Rev., 1980, 80, 63–97 CrossRef CAS.
- (a) 1,3-Dipolar Cycloaddition Chemistry, Vol. 1 (ed. A. Padwa), Wiley, New York, 1984 Search PubMed; (b) R. Huisgen, in Advances in Cycloaddition, Vol. 1, (ed.: D. P. Curran), JAI Press Inc., Greenwich, 1988, p1–32 Search PubMed; (c) P. Deshong; S. W. Lander; J. M. Leginus Jr.; C. M. Dicken in Advances in Cycloaddition, Vol. 1, (ed.: D. P. Curran), JAI Press Inc., Greenwich, 1988, pp. 87–128 Search PubMed; (d) A. Padwa in Advances in Cycloaddition, Vol. 2, (ed.: D. P. Curran), JAI Press Inc., Greenwich, 1990, pp. 1–91 Search PubMed; (e) W. Carruthers in Cycloaddition Reactions in Organic Synthesis, Pergamon, Oxford, 1990 Search PubMed; (f) A. Padwa in Comprehensive Organic Synthesis, Vol. 4 (ed.: B. M. Trost; I. Flemming), Wiley, New York, 1991 Search PubMed; (g) P. A. Wade in Comprehensive Organic Synthesis, Vol. 4(ed.: B. M. Trost; I. Flemming), Wiley, New York, 1991 Search PubMed; (h) R. Huisgen in 1,3-Dipolar Cycloaddition Chemistry, Vol. 1 (ed.: A. Padwa), Wiley, New York, 1984, pp. 1–176 Search PubMed; (i) S. Kanemasa, Synlett, 2002, 1371 CrossRef CAS; (j) K. V. Gothelf in Cycloaddition Reactions in Organic Synthesis (ed.: S. Kobayashi; K. A. Jorgensen), Wiley-VCH, Weinheim, 2002, pp. 211–248 Search PubMed; (k) S. Kanemasa in Cycloaddition Reactions in Organic Synthesis (ed.: S. Kobayashi; K. A. Jorgensen), Wiley-VCH, Weinheim, 2002, pp. 249–300 Search PubMed.
- (a) For a pertinent review, see: M. Lautens, W. Klute and W. Tam, Chem. Rev., 1996, 96, 49–92 Search PubMed, for more recent examples see (b) P. A. Wender and T. J. Williams, Angew. Chem., Int. Ed., 2002, 41, 4550 CrossRef CAS; (c) P. A. Wender, G. G. Gamber, R. D. Hubbard and L. Zhang, J. Am. Chem. Soc., 2002, 124, 2876 CrossRef CAS; (d) P. A. Wender and N. M. Deschamps, Angew. Chem., Int. Ed., 2003, 42, 1853 CrossRef CAS; (e) P. A. Wender, G. G. Gamber, R. D. Hubbard, S. M. Pham and L. Zhang, J. Am. Chem. Soc., 2005, 127, 2836 CrossRef CAS; (f) P. A. Wender, M. P. Croatt and N. M. Deschamps, Angew. Chem., Int. Ed., 2006, 45, 2459 CrossRef CASand references therein.
- (a) For pioneering contributions see: R. Huisgen, Angew. Chem., Int. Ed. Engl., 1963, 2, 565–598 Search PubMedfor early reviews on nitrones, see: (b) L. I. Smith, Chem. Rev., 1938, 38, 193–285 CrossRef; (c) J. Hamer and A. Macaluso, Chem. Rev., 1964, 64, 473–495 CrossRef CASsee also (d) P. Merino in Science of Synthesis, Vol. 27 (ed.: A. Padwa), Thieme, Stuttgart, 2004, pp. 511–580 Search PubMed; (e) J. J. Tufariello in 1,3-Dipolar Cycloaddition Chemistry, Vol. 2 (ed.: A. Padwa), Wiley, New York, 1984, pp. 83–168 Search PubMed; (f) M. Frederickson, Tetrahedron, 1997, 53, 403–425 CrossRef CAS; (g) K. V. Gothelf and K. A. Jørgensen, Chem. Rev., 1998, 98, 863–909 CrossRef CAS; (h) R. C. F. Jones; J. N. Martin in Synthetic Applications of 1,3-Dipolar Cycloaddition Chemistry Toward Heterocycles and Natural Products, The Chemistry of Heterocyclic Compounds, Vol. 59 (ed.: A. Padwa; W. H. Pearson), Wiley, New York, 2002, pp. 1–81 Search PubMed.
- (a) I. S. Young and M. A. Kerr, Angew. Chem., Int. Ed., 2003, 42, 3023 CrossRef CAS; (b) M. D. Ganton and M. A. Kerr, J. Org. Chem., 2004, 69, 8554 CrossRef CAS; (c) A. Karadeolian and M. A. Kerr, J. Org. Chem., 2007, 72, 10251 CrossRef CAS; (d) M. P. Sibi, Z. Ma and C. P. Jasperse, J. Am. Chem. Soc., 2005, 127, 5764 CrossRef CAS; (e) Y.-B. Kang, X.-L. Sun and Y. Tang, Angew. Chem., Int. Ed., 2007, 46, 3918 CrossRef CAS.
- A. H. Stoll and S. B. Blakey, J. Am. Chem. Soc., 2010, 132, 2108 CrossRef CAS.
- (a) B. M. Trost, Angew. Chem., 1986, 98, 1 CAS; (b) M. Harmata, Adv. Cycloaddit., 1997, 4, 41 Search PubMed; (c) M. Harmata, Tetrahedron, 1997, 53, 6235 CrossRef CAS; (d) E. Nakamura and S. Yamago, Acc. Chem. Res., 2002, 35, 867 CrossRef CAS; (e) S. Yamago and E. Nakamura, Org. React., 2002, 61, 1 CAS.
- (a) H. M. R. Hoffmann, Angew. Chem., Int. Ed. Engl., 1984, 23, 1 CrossRef; (b) C. B. W. Stark, U. Eggert and H. M. R. Hoffmann, Angew. Chem., Int. Ed., 1998, 37, 1266 CrossRef CAS; (c) C. B. W. Stark, S. Pierau, R. Wartchow and H. M. R. Hoffmann, Chem.–Eur. J., 2000, 6, 684 CrossRef CAS.
- (a) A. S. Kende and H. Huang, Tetrahedron Lett., 1997, 38, 3353 CrossRef CAS; (b) S.-J. Jin, J. R. Choi, J. Oh, D. Lee and J. K. Cha, J. Am. Chem. Soc., 1995, 117, 10914 CrossRef CAS; (c) J. Oh, J. Lee and S.j. Jin, Tetrahedron Lett., 1994, 35, 3449 CrossRef CAS.
- G. Prie, N. Prevost, H. Twin, S. A. Fernandes, J. F. Hayes and M. Shipman, Angew. Chem., Int. Ed., 2004, 43, 6517 CrossRef CAS.
- (a) A. R. Thornton and S. B. Blakey, J. Am. Chem. Soc., 2008, 130, 5020 CrossRef CAS; (b) A. R. Thornton, V. I. Martin and S. B. Blakey, J. Am. Chem. Soc., 2009, 131, 2434 CrossRef CAS.
- C. G. Espino, P. M. Wehn, J. Chow and J. Du Bois, J. Am. Chem. Soc., 2001, 123, 6935 CrossRef CAS.
- The relative stereochemistry was proven by X-ray crystallography or derivatization followed by X-ray crystallography. See ESI† for details.
- T. Kurokawa, M. Kim and J. Du Bois, Angew. Chem., Int. Ed., 2009, 48, 2777 CrossRef CAS.
- In a related study, Robertson and co-workers also comment on the timing of the bond forming events (a) G. C. Feast, L. W. Page and J. Robertson, Chem. Commun., 2010, 46, 2835 RSC; (b) J. Robertson, G. C. Feast, L. V. White, V. A. Steadman (née Doughty) and T. D. W. Claridge, Org. Biomol. Chem., 2010, 8, 3060 RSC.
- M. T. Reetz and J. Rheinheimer, J. Org. Chem., 1986, 51, 5465 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details and structural proofs for all new compounds. CCDC reference numbers 750090 and 750091. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c0sc00375a |
| This journal is © The Royal Society of Chemistry 2011 |