High-resolution and sensitivity through-bond correlations in ultra-fast magic angle spinning (MAS) solid-state NMR†
Ivano
Bertini
a,
Lyndon
Emsley
b,
Isabella C.
Felli
a,
Ségolène
Laage
b,
Anne
Lesage
*b,
Józef R.
Lewandowski
b,
Alessandro
Marchetti
bc,
Roberta
Pierattelli
a and
Guido
Pintacuda
*b
aDepartment of Chemistry “Ugo Schiff” and Magnetic Resonance Center (CERM), University of Florence, Via Luigi Sacconi 6, 50019, Sesto Fiorentino, Firenze, Italy
bUniversité de Lyon, CNRS/ENS Lyon/UCB-Lyon 1, Centre RMN à Très Hauts Champs, 5 rue de la Doua, 69100, Villeurbanne, France. E-mail: anne.lesage@ens-lyon.fr; guido.pintacuda@ens-lyon.fr; Fax: +33 4 78 89 67 61; Tel: +33 4 26 23 38 88
cScuola Normale Superiore, Piazza dei Cavalieri 7, 56126, Pisa, Italy
First published on 15th November 2010
Abstract
We introduce a new experiment, which makes use of Spin State Selective manipulations to perform sensitive and resolved through-bond correlations in organic and biological solids at high-fields and under ultra-fast MAS. The scheme is the shortest and most sensitive through-bond correlation method introduced so far in solids, yields resolved fingerprints of uniformly 13C-labeled biomolecules, and constitutes a tool to highlight slight static structural disorder around crystallographically equivalent molecules in microcrystalline samples.
In recent years, NMR has progressed greatly as a structural and dynamic probe of biomolecular solids.1–3 Two- or three-dimensional homonuclear and heteronuclear correlation experiments are at the heart of such studies, for example correlating 13C, 15N resonances of uniformly 13C- and 15N-labeled proteins.4–6 One of the key obstacles to further progress is that the efficiency of such correlation experiments is usually limited, especially when using the through-bond J coupling as the interaction to generate correlations.7
The recent development of MAS probes capable of achieving so-called fast (>30 kHz) and ultra-fast (>50 kHz) spinning rates has been revolutionizing this field,8–15 opening up several new perspectives in the analysis of a larger range of systems in many areas of modern biology.16–19 Under ultra-fast MAS, heteronuclear decoupling is efficiently performed with low-power irradiation.20–23 Therefore, long coherence lifetimes can be obtained without the negative effects of strong radio-frequency (RF) fields,24 even without the need of extensive deuteration.25 This enables both an increase in resolution, by allowing longer acquisition times in direct and indirect dimensions of multidimensional correlations, and in sensitivity, by shortening the interscan delays, without unwanted heating and subsequent deterioration of the sample.11,26 In this context, the exploitation of J-transfer based correlation experiments,7,24,27–35 where long evolution delays are necessary for creating and refocusing coherences between neighboring spins, becomes increasingly pertinent.36
Fig. 1a demonstrates how J-scalar transfer efficiency grows at ultra-fast MAS as a result of the longer coherence lifetimes. This figure compares the performance of cross-polarization (CP) transfers to CP-refocused-INADEQUATE experiments37,38 for the CO resonances of the microcrystalline protein domain GB1.39 A significant increase of the coherence lifetimes of both the CO and Cα spins is observed moving from 10 kHz MAS (e.g. coherence lifetimes T2′ of 12.8 ms for the CO spins under 80 kHz SPINAL-6440 decoupling), to 60 kHz MAS under low-power decoupling (e.g. coherence lifetimes T2′ of 86 ms for the CO spins under 15 kHz swept low-power TPPM (slpTPPM) decoupling41). Consequently, the efficiency of the through-bond transfers is significantly increased from 15 to 45%, thus rendering this kind of experiment a competitive alternative to dipolar-based schemes not only in perdeuterated molecules,25,42 but also in fully protonated substrates.
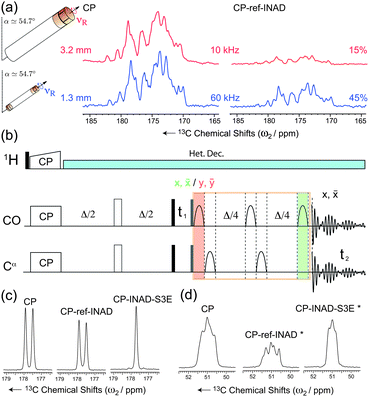 | ||
| Fig. 1 (a) Efficiency of the DQ-INADEQUATE experiment (right) as compared to CP (left) for microcrystalline GB1 at νR = 10 kHz MAS (80 kHz SPINAL-6440) and νR = 60 kHz MAS (15 kHz slpTPPM41). Percentages refer to relative peak heights. (b) Pulse sequence for the band-selective refocused INADEQUATE-S3E experiment. The bell shapes represent the band-selective π pulses and the delay Δ/4 is set to 1/8J. For each t1 increment, two FIDs are recorded, with a CO selective π pulse placed either at the end (in green) or at the beginning (in red) of the refocusing block, together with a suitable phase cycle of the second π/2 hard pulse (grey rectangle) and the receiver. (c–d) Relative efficiency of CP (left), INADEQUATE (center), and INADEQUATE-S3E (right) recorded on CO (c) and Cα (d) resonances of fully 13C-labeled L-Alanine, at 60 kHz MAS and low-power slpTPPM (ω1H/2π∼νR/4 = 13.6 kHz according to pulse calibration). The Cα INADEQUATE signals (*) were extracted from selective 2D INADEQUATE experiments, so that only CO magnetization was excited at the CP step. | ||
In parallel, resolution is also a key barrier to opening the door to solid-state NMR towards increasingly complex biological samples. A variety of J-decoupling techniques have recently been proposed to remove the scalar splittings between neighboring 13C spins,43,44 which significantly affect the spectral resolution in fully 13C-labeled systems.24,31,45–49
Here we introduce a new experiment which simultaneously improves the performance of through-bond transfers and accomplishes virtual homonuclear J-decoupling. As illustrated in Fig. 1b, this is achieved by incorporating the S3E scheme43,44,49 (Spin State Selective Excitation) into the INADEQUATE after the evolution of the DQ coherences.
This block allows recovery of in-phase signals efficiently by partial inter-conversion between in-phase and antiphase components, without the need of complete refocusing of antiphase coherences. This requires half of the time (1/4J i.e. 4.5 ms for a JCOCα = 55 Hz) of the reconversion block for corresponding refocused INADEQUATE, minimizing the duration of the pulse sequence and the relative losses due to transverse decay. Additionally, it provides the two individual doublet components separately, which can then be combined into a single, more intense signal simultaneously removing the J-splitting and increasing the overall signal to noise by a further factor 1.4.44 It is noteworthy that, in contrast to the previous implementation in NCO or NCA experiments, where the S3E element was appended to the end of the pulse sequence,49 the S3E block here replaces the conventional refocusing periods of the INADEQUATE experiments. This pulse scheme represents therefore the shortest in-phase J-based correlation experiment designed so far, which results in minimal loss of sensitivity through transverse dephasing. Finally, this scheme mutually decouples the two spins from each other, leading to an increase of resolution and sensitivity on both spins. As illustrated in Fig. 1c and d for [U-13C]-L-Alanine, sensitivity gains (as measured by the relative peak heights) of factors of 1.4 (CO) and 2.0 (Cα) are observed when the S3E block is used instead of the conventional refocusing period in an INADEQUATE experiment. Interestingly, in this new experiment, the CO peak height after the through-bond transfer is actually more intense than that of each component of the doublet acquired straight after CP. The scheme lends itself beautifully to the investigation of large biological solids at high-fields and ultra-fast MAS. Fig. 2a shows the carbonyl region of a 2D INADEQUATE-S3E experiment recorded on microcrystalline oxidized superoxide dismutase (SOD), a dimeric paramagnetic enzyme of 32 kDa50 recorded with 60 kHz MAS on an 850 MHz spectrometer. The incorporation of the S3E block yields a gain in resolution, removing all the 55 Hz one-bond JCOCα couplings, and in sensitivity up to a factor 1.8 (see the ESI† for the sensitivity gains for all of the resolved correlations). As a consequence of the shorter refocusing period, for most of the signals the sensitivity gain exceeds the limit of 1.4, provided by the simple combination of the two doublet components. This is illustrated by the traces in Fig. 2b. Notably, conventional refocused INADEQUATE spectra of comparable S/N, with dramatically reduced resolution, can typically be acquired under slow MAS, in over double the time with a 10-fold larger amount of sample, see the ESI†.
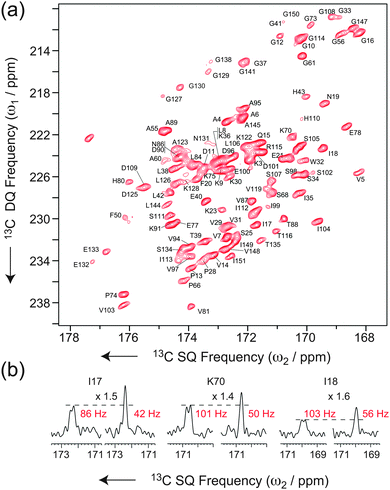 | ||
| Fig. 2 (a) Carbonyl region of a band-selective INADEQUATE-S3E recorded at νR = 60 kHz MAS on dimeric oxidized SOD (see ESI† for details). (b) Traces of 2D INADEQUATE spectra acquired using the conventional refocusing period (left) and the S3E block (right). The gains in sensitivity are, respectively, 1.5, 1.4 and 1.6. | ||
Besides providing an unambiguous identification of resonances, through-bond correlation methods in solids are tools to investigate structural disorder.36
For example, in disordered systems like glasses, natural polymers or silica compounds, the INADEQUATE experiment is well known to provide resolved correlations between the chemical shift distributions of adjacent nuclei.51,52Fig. 3a shows the application of the INADEQUATE-S3E to N,N-bis(diphenylphosphino)-N-((S)-α-methylbenzyl)amine, a slightly disordered crystalline organic compound.53,54 Here high-resolution correlations between individual chemical shift distributions can be extracted for each of four 31P–N-31P pairs corresponding to the four conformationally distinct residues in the crystal structure. In turn, this provides an accurate sampling of the local conformations, as, for example, structures generated along low-frequency vibrational modes can reproduce these correlations.53
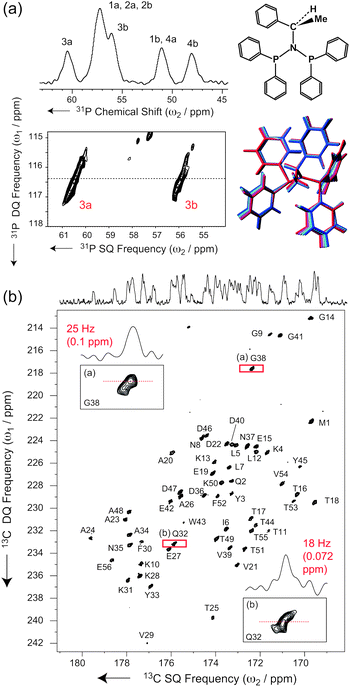 | ||
| Fig. 3 (a) 31P-1H CPMAS and INADEQUATE-S3E spectra of N,N-bis(diphenylphosphino)-N-((S)-α-methylbenzyl)amine recorded at 11.7 T and at a MAS frequency of νR = 60 kHz, under low-power slpTPPM 1H decoupling (ω1H/2π∼νR/4 = 16 kHz according to flip pulse calibration). See ESI† for further details. For this disordered molecule, the planar formula is shown, together with a structure bundle generated from low-frequency vibrational modes and compatible with the observed 2D 31P-lineshapes (see ref. 53). (b) Carbonyl region of a band-selective INADEQUATE-S3E spectrum of microcrystalline GB139 recorded at νR = 60 kHz MAS on a 1000 MHz Bruker Avance III spectrometer (see ESI† for details). | ||
The higher resolution provided by the S3E implementation of the INADEQUATE experiment under ultra-fast MAS is a new tool to highlight this same kind of effect in a microcrystalline protein, and to potentially observe the detailed structure of the correlation peaks in analogy to disordered organic solids. The corresponding spectrum recorded on a well-defined polymorph of microcrystalline protein domain GB1 (form A)55 indeed displays lineshapes with peculiar fine structures, which are the signature of correlated distributions of isotropic shift frequencies associated with slight static structural disorder around each crystallographically equivalent molecule. In addition, as compared to the inhomogeneously broadened signal, in this spectrum, individual cross sections feature increased resolution (Fig. 3b), with a further narrowing of the linewidth.
In summary, we have introduced a new NMR experiment, which performs sensitive and resolved through-bond correlations in solids under ultra-fast MAS. This is expected to become an important tool for the study of challenging biological systems in the solid-state, for which obtaining resolved fingerprints is a key step for any structural and biophysical characterization.
Acknowledgements
We acknowledge support from the Agence Nationale de la Recherche (ANR 08-BLAN-0035-01), from Ente Cassa di Risparmio di Firenze, from Egide (programme Galilée 22397RJ), from the Università Italo-Francese (Programma Galileo 2009/2010) and from Joint Research Activity and Access to Research Infrastructures activity in the 6th Framework Program of the EC (RII3-026145, EU-NMR). We thank Dr H. J. Sass and Prof. S. Grzesiek for providing a 13C, 15N labeled sample of GB1, Dr M. Bardet (Grenoble) for providing the phosphorous sample and Dr S. Cadars (Orléans) for stimulating discussions. J.R.L. was supported by EU IRG (PIRG03- GA-2008-231026).Notes and references
- M. Baldus, Curr. Opin. Struct. Biol., 2006, 16, 618–623 CrossRef CAS.
- A. E. McDermott and T. Polenova, Curr. Opin. Struct. Biol., 2007, 17, 617–622 CrossRef CAS.
- A. Böckmann, Angew. Chem., Int. Ed., 2008, 47, 6110–6113 CrossRef.
- S. Luca, H. Heise and M. Baldus, Acc. Chem. Res., 2003, 36, 858–865 CrossRef CAS.
- A. E. McDermott, Curr. Opin. Struct. Biol., 2004, 14, 1–8 CrossRef.
- L. J. Sperling, D. A. Berthold, T. L. Sasser, V. Jeisy-Scott and C. M. Rienstra, J Mol Biol, 2010, 399, 268–282 CrossRef CAS.
- A. Lesage, “Indirect Coupling and Connectivity” in Encyclopedia of Magnetic Resonance, ed. R. K. Harris and R. Wasylishen, John Wiley: Chichester, 2008 DOI:10.1002/9780470034590.emrstm1012.
- D. H. Zhou, J. J. Shea, A. J. Nieuwkoop, W. T. Franks, B. J. Wylie, C. Mullen, D. Sandoz and C. M. Rienstra, Angew. Chem., Int. Ed., 2007, 46, 8380–8383 CrossRef CAS.
- D. H. Zhou, G. Shah, M. Cormos, C. Mullen, D. Sandoz and C. M. Rienstra, J. Am. Chem. Soc., 2007, 129, 11791–11792 CrossRef CAS.
- S. Laage, A. Marchetti, J. Sein, R. Pierattelli, H. J. Sass, S. Grzesiek, A. Lesage, G. Pintacuda and L. Emsley, J. Am. Chem. Soc., 2008, 130, 17216–17217 CrossRef CAS.
- S. Laage, J. Sachleben, S. Steuernagel, R. Pierattelli, G. Pintacuda and L. Emsley, J. Magn. Reson., 2008, 133–141.
- D. H. Zhou, G. Shah, C. Mullen, D. Sandoz and C. M. Rienstra, Angew. Chem., Int. Ed., 2009, 48, 1253–1256 CrossRef CAS.
- A. Lange, I. Scholz, T. Manolikas, M. Ernst and B. H. Meier, Chem. Phys. Lett., 2009, 468, 100–105 CrossRef CAS.
- V. Vijayan, J. P. Demers, J. Biernat, E. Mandelkow, S. Becker and A. Lange, ChemPhysChem, 2009, 10, 2205–2208 CrossRef CAS.
- I. Bertini, L. Emsley, M. Lelli, C. Luchinat, J. Mao and G. Pintacuda, J. Am. Chem. Soc., 2010, 132, 5558–5559 CrossRef CAS.
- C. Wasmer, A. Lange, H. Van Melckebeke, A. B. Siemer, R. Riek and B. H. Meier, Science, 2008, 319, 1523–1526 CrossRef CAS.
- P. Turano, D. Lalli, I. C. Felli, E. C. Theil and I. Bertini, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 545–550 CrossRef CAS.
- S. D. Cady, K. Schmidt-Rohr, J. Wang, C. S. Soto, W. F. DeGrado and M. Hong, Nature, 2010, 463, 689–692 CrossRef CAS.
- E. Barbet-Massin, S. Ricagno, J. R. Lewandowski, S. Giorgetti, V. Bellotti, M. Bolognesi, L. Emsley and G. Pintacuda, J. Am. Chem. Soc., 2010, 132, 5556–5557 CrossRef CAS.
- M. Ernst, A. Samoson and B. H. Meier, Chem. Phys. Lett., 2001, 348, 293–302 CrossRef CAS.
- M. Ernst, A. Samoson and B. H. Meier, J. Magn. Reson., 2003, 163, 332–339 CrossRef CAS.
- M. Ernst, M. A. Meier, T. Tuherm, A. Samoson and B. H. Meier, J. Am. Chem. Soc., 2004, 126, 4764–4765 CrossRef CAS.
- M. Kotecha, N. P. Wickramasinghe and Y. Ishii, Magn. Reson. Chem., 2007, 45, S221–S230 CrossRef CAS.
- L. L. Chen, J. M. Kaiser, J. F. Lai, T. Polenova, J. Yang, C. M. Rienstra and L. J. Mueller, Magn. Reson. Chem., 2007, 45, S84–S92 CrossRef CAS.
- M. Hologne, V. Chevelkov and B. Reif, Prog. Nucl. Magn. Reson. Spectrosc., 2006, 48, 211–232 CrossRef CAS.
- N. P. Wickramasinghe, S. Parthasarathy, C. R. Jones, C. Bhardwaj, F. Long, M. Kotecha, S. Mehboob, L. W. M. Fung, J. Past, A. Samoson and Y. Ishii, Nat. Methods, 2009, 6, 215–218 CrossRef CAS.
- R. Verel, J. D.v. Beek and B. H. Meier, J. Magn. Reson., 1999, 140, 300–303 CrossRef CAS.
- G. De Paepe, N. Giraud, A. Lesage, P. Hodgkinson, A. Böckmann and L. Emsley, J. Am. Chem. Soc., 2003, 125, 13938–13939 CrossRef CAS.
- L. L. Chen, R. A. Olsen, D. W. Elliott, J. M. Boettcher, D. H. H. Zhou, C. M. Rienstra and L. J. Mueller, J. Am. Chem. Soc., 2006, 128, 9992–9993 CrossRef CAS.
- L. Chen, J. M. Kaiser, T. Polenova, J. Yang, C. M. Rienstra and L. J. Mueller, J. Am. Chem. Soc., 2007, 129, 10650–10651 CrossRef CAS.
- R. Verel, T. Manolikas, A. B. Siemer and B. H. Meier, J. Magn. Reson., 2007, 184, 322–329 CrossRef CAS.
- R. Linser, U. Fink and B. Reif, J. Magn. Reson., 2008, 193, 89–93 CrossRef CAS.
- A. Loquet, S. Laage, C. Gardiennet, B. Elena, L. Emsley, A. Böckmann and A. Lesage, J. Am. Chem. Soc., 2008, 130, 10625–10632 CrossRef CAS.
- D. Lee, J. Struppe, D. W. Elliott, L. J. Mueller and J. J. Titman, Phys. Chem. Chem. Phys., 2009, 11, 3547–3553 RSC.
- Y. Tian, L. L. Chen, D. Niks, J. M. Kaiser, J. F. Lai, C. M. Rienstra, M. F. Dunn and L. J. Mueller, Phys. Chem. Chem. Phys., 2009, 11, 7078–7086 RSC.
- A. Lesage, Phys. Chem. Chem. Phys., 2009, 11, 6876–6891 RSC.
- A. Lesage, M. Bardet and L. Emsley, J. Am. Chem. Soc., 1999, 121, 10987–10993 CrossRef CAS.
- S. Cadars, J. Sein, L. Duma, A. Lesage, T. N. Pham, J. H. Baltisberger, S. P. Brown and L. Emsley, J. Magn. Reson., 2007, 188, 24–34 CrossRef CAS.
- W. T. Franks, D. H. Zhou, B. J. Wylie, B. G. Money, D. T. Graesser, H. L. Frericks, G. Sahota and C. M. Rienstra, J. Am. Chem. Soc., 2005, 127, 12291–12305 CrossRef CAS.
- B. M. Fung, A. K. Khitrin and K. Ermolaev, J. Magn. Reson., 2000, 142, 97–101 CrossRef CAS.
- J. R. Lewandowski, J. Sein, H. J. Sass, S. Grzesiek, M. Blackledge and L. Emsley, J. Am. Chem. Soc., 2010, 132, 8252–8254 CrossRef CAS.
- P. Schanda, M. Huber, R. Verel, M. Ernst and B. H. Meier, Angew. Chem., Int. Ed., 2009, 48, 9322–9325 CrossRef CAS.
- W. Bermel, I. Bertini, I. C. Felli, M. Matzapetakis, R. Pierattelli, E. C. Thiel and P. Turano, J. Magn. Reson., 2007, 188, 301–310 CrossRef CAS.
- W. Bermel, I. C. Felli, R. Kümmerle and R. Pierattelli, Concepts Magn. Reson. A, 2008, 32A, 183–200 CrossRef CAS.
- L. Duma, S. Hediger, B. Brutscher, A. Böckmann and L. Emsley, J. Am. Chem. Soc., 2003, 125, 11816–11817 CrossRef.
- V. Chevelkov, Z. J. Chen, W. Bermel and B. Reif, J. Magn. Reson., 2005, 172, 56–62 CrossRef CAS.
- T. I. Igumenova and A. E. McDermott, J. Magn. Reson., 2005, 175, 11–20 CrossRef CAS.
- I. Scholz, S. Jehle, P. Schmieder, M. Hiller, F. Eisenmenger, H. Oschkinat and B. J. van Rossum, J. Am. Chem. Soc., 2007, 129, 6682–6683 CrossRef CAS.
- S. Laage, A. Lesage, L. Emsley, I. Bertini, I. C. Felli, R. Pierattelli and G. Pintacuda, J. Am. Chem. Soc., 2009, 131, 10816 CrossRef CAS.
- G. Pintacuda, N. Giraud, R. Pierattelli, A. Böckmann, I. Bertini and L. Emsley, Angew. Chem., Int. Ed., 2007, 46, 1079–1082 CrossRef CAS.
- S. Cadars, A. Lesage and L. Emsley, J. Am. Chem. Soc., 2005, 127, 4466–4476 CrossRef CAS.
- D. Sakellariou, S. P. Brown, A. Lesage, S. Hediger, M. Bardet, C. A. Meriles, A. Pines and L. Emsley, J. Am. Chem. Soc., 2003, 125, 4376–4380 CrossRef CAS.
- S. Cadars, A. Lesage, C. J. Pickard, P. Sautet and L. Emsley, J. Phys. Chem. A, 2009, 113, 902–911 CrossRef CAS.
- S. Cadars, A. Lesage, M. Trierweiler, L. Heux and L. Emsley, Phys. Chem. Chem. Phys., 2007, 9, 92–103 RSC.
- H. L. F. Schmidt, L. J. Sperling, Y. G. Gao, B. J. Wylie, J. M. Boettcher, S. R. Wilson and C. A. Rienstra, J. Phys. Chem. B, 2007, 111, 14362–14369 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental section, scalar and dipolar correlation spectra of SOD, sensitivity gains for the resolved peaks in the carbonyl regions of SOD, INADEQUATE-S3E spectra of N,N-bis(diphenylphosphino)-N-((S)-α-methylbenzyl)amine, pulse schemes with full phase cycles. See DOI: 10.1039/c0sc00397b |
| This journal is © The Royal Society of Chemistry 2011 |