In(III)-pybox complex catalyzed enantioselective Mukaiyama aldol reactions between polymeric or hydrated glyoxylates and enolsilanes derived from aryl ketones†
Jun-Feng
Zhao
,
Boon-Hong
Tan
and
Teck-Peng
Loh
*
Division of Chemistry and Biological Chemistry, School of Physical and Mathematical Sciences, Nanyang Technological University, Singapore 637616. E-mail: teckpeng@ntu.edu.sg; Fax: +65 6791 1961; Tel: +65 6316 8899
First published on 28th October 2010
Abstract
A highly efficient In(III)-pybox complex catalyzed enantio-selective Mukaiyama aldol reaction between polymeric or hydrated glyoxylates and enolsilanes derived from aryl ketones is described. Excellent enantioselectivities, diastereoselectivities and yields were obtained with a broad substrate scope. The mild reaction conditions and simple operation make this methodology very practical.
The aldol reaction offers one of the most efficient access to the versatile β-hydroxyl carbonyl compounds, which are important building blocks for biologically active molecules.1 It is not surprising that the asymmetric aldol reaction has attracted much attention in recent years. Among the strategies elaborated in the asymmetric aldol reactions, chiral Lewis-acid catalyzed asymmetric Mukaiyama aldol reaction has been developed into a useful tool for organic chemists.2 Although asymmetric direct aldol reactions3 have developed very rapidly in the past decades, the asymmetric Mukaiyama aldol still plays an important role in organic synthesis. For example, the organocatalyzed asymmetric direct aldol reaction of poorly reactive aryl ketones represents a challenging topic4 albeit exciting results having been achieved in metal complexes such as dinuclear Zn catalysts5 and (S,S)-Zn–Zn-linked-BINOL complex6 catalyzed asymmetric direct aldol reaction. However, the chiral Lewis acids catalyzed asymmetric Mukaiyama aldol of aryl ketones, in which preformed aryl enol silanes were used as aldol donors, can avoid the disadvantage caused by the low reactivity of aryl ketone.7
On the other hand, the enantioselective Mukaiyama aldol reaction of 1,2-dicarbonyl compounds has emerged as an efficient strategy to construct enantiomerically enriched β-hydroxy ketones and esters, which are frequently observed in numerous biologically active molecules.8 A number of chiral catalysts have been developed for the asymmetric Mukaiyama aldol reaction of α-keto esters9 and glyoxylates.10 To our surprise, despite the large number of chiral catalyst capable of mediating the asymmetric aldol reaction of glyoxylates and aryl ketones, individually, none of them are effective for the asymmetric aldol reaction between aryl ketones and glyoxylates except for Evans' Sc(III)-pybox complex.11 Unfortunately, Evans' catalyst system only worked for substituted steric bulky aryl ketones12 and strictly anhydrous reaction conditions were required. Therefore, the asymmetric aldol reaction between glyoxylates and aryl ketones, which can generate the carbonyl precursor of (R)-ethyl 2-hydroxy-4-phenylbutyrate (HPB ester),13 a key intermediate of a series of commercially available Angiotensin-Converting Enzyme (ACE) inhibitors,14 still remained as a formidable challenge. Currently enantioselective synthesis of this compound relies upon asymmetric aza-ene type reaction of enecarbamate,15 in which the synthesis of enecarbamate is very tedious. Herein, we report a highly efficient asymmetric In(III)-pybox complex catalyzed Mukaiyama aldol reaction between polymeric or hydrated glyoxylate and enolsilanes derived from aryl ketones to give enantioenriched β-hydroxy ketones in excellent yields and enantioselectivities under mild reaction conditions.
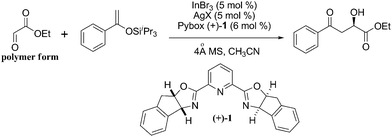
In the course of our effort to develop the application of chiral indium complex16 in asymmetric synthesis, we demonstrated that In(III)-pybox complexes, formed in situ from commercially available In(III) salts and pybox ligands, are efficient catalysts for asymmetric carbonyl-ene reaction of polymeric glyoxylates and trifluoropyruvates.17 This may be attributed to glyoxylate and trifluoropyruvate, both being bidentate substrates, coordinating to the cationic In(III) centre to form a facially discriminated complex, which is crucial for inducing high enantioselectivity. We hypothesized that this strategy could also be applied to the challenging asymmetric Mukaiyama aldol reaction beween glyoxylate and aryl enol silanes, which are similar to α-methylstyrene.
Initially, we chose polymeric ethyl glyoxylate (50% in toluene) and acetophenone-derived enolsilane as the model substrates to optimize reaction conditions (for details of screening of different Lewis acids and condition optimization, see ESI†). It is notable that among the evaluated Lewis acids, only In(III) based Lewis acids are effective for this reaction. The solvent, ligand, temperature, additive, catalyst loading and counterion effect were all examined. Polar solvents such as CH3CN and THF proved to be better than non-polar solvents. Pybox (+)-1 is the best choice of ligand and box ligands are ineffective for this transformation. With decreasing temperature, the enantioselectivity was found to increase. In addition, due to the significant counter-ion effect of In(III)-pybox complex observed in the asymmetric carbonyl-ene reactions, the counter-ion effect also had been evaluated in the current study as shown in Table 1. Based on the optimization studies, we found that the combination of 5 mol% of InBr3, 5 mol% of AgSbF6 and 6 mol% of pybox (+)-1 with CH3CN as the solvent in the presence of 4 Å molecular sieves18 was the best catalytic system.‡
| Entry | AgX (5 mol %) | T/°C | t/h | Yieldb (%) | eec (%) (R) |
|---|---|---|---|---|---|
| a The reactions were carried out on a 0.50 mmol scale of enolsilane with 2 equiv of ethyl glyoxylate (1 mmol) in acetonitrile (CH3CN). b Isolated yield. c ee Values were determined by chiral stationary phase HPLC analysis. The absolute configuration of the major products is R, assigned by comparing the optical rotation with the literature.13 | |||||
| 1 | AgSbF6 | r.t | 2 | 97 | 77 |
| 2 | AgPF6 | r.t | 4 | 95 | 77 |
| 3 | AgBF4 | r.t | 2 | 96 | 72 |
| 4 | AgClO4 | r.t | 4 | 95 | 73 |
| 5 | AgOAc | r.t | 24 | — | nd |
| 6 | AgSbF6 | −20 | 19 | 89 | 87 |
| 7 | AgSbF 6 | −40 | 72 | 91 | 89 |
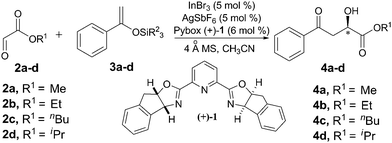
In our preliminary studies, consistent with Evans' results, we also observed that this reaction is sensitive to the steric effect of reactants. Increasing steric bulk of the substrate is favorable for attaining higher enantioselectivity (unpublished results). These factors promoted us to investigate the steric effect of glyoxylate esters, as well as enolsilanes systematically, with the hope of getting better enantioselectivity, and the results are listed in Table 2. Firstly, acetophenone derived enolsilanes with different size of silane groups were evaluated at room temperature. It is clear that the enantioselectivity increased steadily from 69 to 77% when the silane group changed from small TMS (trimethylsilyl) to bulky TIPS (triisopropylsilyl) (Table 2, entries 1–4). Next, a series of glyoxylate esters with different ester groups were prepared according to the reported method19 and used as their hydrated form without further distillation. Keeping TIPS as the silane group, similar increasing trend in enantioselectivity was observed with increasing size of glyoxylate ester group (Table 2, entries 4–7). The highest ee (89%) could be achieved when isopropyl glyoxylate ester and TIPS enolsilane derived from acetophenone, both of which contain the bulkiest isopropyl group, were reacted under the standard conditions at room temperature (Table 2, entry 7). The results summarized in Table 2 supported our hypothesis that increasing steric bulk of both enolsilanes and glyoxylate esters will lead to increase in the enantioselectivity. Further improvement of enantioselectivity was observed when the reaction was carried out at low temperature (Table 2, entries 8–10). The optimal temperature to afford the best results in terms of yield and enantioselectivity is −20 °C (Table 2, entry 9), while too low a temperature has a detrimental effect on the reaction efficiency (Table 2, entry 10). Interestingly, unlike Evans' strict anhydrous conditions, under which acidic treatment is necessary to transfer the silane ether product to alcohol, the enantioenriched alcohol could be obtained directly from Mukaiyama aldol reaction in our case. This result demonstrated that a small amount of water in this system plays a role in this process.
| Entry | R1 | SiR23 | T/°C | t/h | Yieldb (%) | eec (%) |
|---|---|---|---|---|---|---|
| a The reactions were carried out on a 0.50 mmol scale of 3 with 2 equiv of 2 (1 mmol) in acetonitrile (CH3CN). b Isolated yield. c ee Values were determined by chiral-phase HPLC analysis. The absolute configurations of 4a, 4c and 4d were assigned by analogy to that of 4b. d This reaction is not completed after stirring for 3 days. | ||||||
| 1 | Et | SiMe3 | r.t | 2 | 80 | 69 |
| 2 | Et | SiEt3 | r.t | 2 | 85 | 72 |
| 3 | Et | SiMe2tBu | r.t | 2 | 87 | 66 |
| 4 | Et | SiiPr3 | r.t | 2 | 97 | 77 |
| 5 | Me | SiiPr3 | r.t | 2 | 62 | 43 |
| 6 | Bu | SiiPr3 | r.t | 2 | 92 | 73 |
| 7 | i Pr | SiiPr3 | r.t | 2 | 93 | 89 |
| 8 | i Pr | SiiPr3 | 0 | 12 | 96 | 92 |
| 9 | i Pr | Si i Pr 3 | −20 | 19 | 91 | 96 |
| 10d | i Pr | SiiPr3 | −40 | 72 | 28 | 97 |
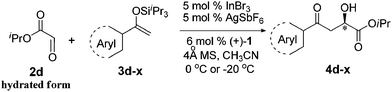
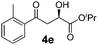
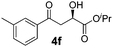
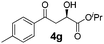
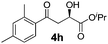
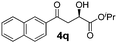
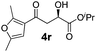
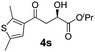
With the optimal conditions in hand, TIPS enolsilanes derived from various aryl ketones were prepared to investigate the substrate scope of this reaction and the results are summarized in Table 3. Arylenolsilanes containing both electron-withdrawing and donating groups on the aromatic ring proceeded smoothly to give the aldol adducts in good to excellent yields and with excellent enantioselectivities, albeit longer reaction times were required for electron-deficient substrates (Table 3, entries 6–10). Interestingly, although the steric hindrance is favorable for the enantioselectivies, too severe steric hindrance in the transition state has a negative effect on the reaction efficiency. For example, the presence of a substituent at the ortho position of enolsilane prolonged the reaction time (up to 3 days) and required higher reaction temperature (0 °C), regardless whether the substituents are electron-neutral (Table 3, entries 2 and 5) or withdrawing (Table 3, entry 6). The disadvantage caused by steric hindrance of orthomethyl or methoxy group may be compensated by the electron-rich nature of the substrates 3n, 3r and 3s (Table 3, entries 11, 15 and 16). These observations demonstrated that both the electronic effect and steric effect play a role in the reaction. It is noteworthy that enolsilanes derived from heterocyclic aryl ketones also worked well to give enantioenriched alcohols which could be elaborated to other useful chemicals (Table 3, entries 15 and 16).
| Entry | Product (4d–x) | T/°C | t/h | Yieldb (%) | eec (%) |
|---|---|---|---|---|---|
| a The reactions were carried out on a 0.50 mmol scale of 3 with 2 equiv of 2 (1 mmol) in acetonitrile (CH3CN). b Isolated yield. c Ee values were determined by chiral-phase HPLC analysis. | |||||
| 1 |
|
−20 | 19 | 91 | 96 |
| 2 |
|
0 | 70 | 81 | 94 |
| 3 |
|
−20 | 22 | 88 | 94 |
| 4 |
|
−20 | 22 | 94 | 96 |
| 5 |
|
0 | 70 | 80 | 95 |
| 6 |
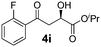
|
0 | 68 | 60 | 93 |
| 7 |
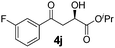
|
0 | 68 | 85 | 93 |
| 8 |
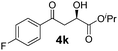
|
−20 | 72 | 71 | 94 |
| 9 |
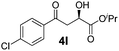
|
−20 | 64 | 82 | 97 |
| 10 |
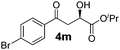
|
−20 | 64 | 85 | 97 |
| 11 |
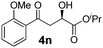
|
−20 | 68 | 83 | 92 |
| 12 |
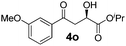
|
−20 | 48 | 85 | 95 |
| 13 |
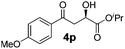
|
−20 | 48 | 88 | 96 |
| 14 |
|
−20 | 64 | 89 | 98 |
| 15 |
|
−20 | 68 | 76 | 90 |
| 16 |
|
−20 | 68 | 92 | 96 |
To further probe the stereoselectivity of this reaction, α-substituted aryl ketone-derived enolsilane 5, which will lead to two chiral centres in the aldol product 6, was reacted under the standard reaction conditions and excellent diastereoselectivity (dr = 97![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3) as well as enantioselectivity (98% ee) could be obtained, with the syn diastereomer as the major product (Scheme 1).
:3) as well as enantioselectivity (98% ee) could be obtained, with the syn diastereomer as the major product (Scheme 1).
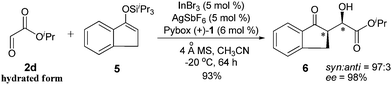 | ||
| Scheme 1 | ||
In summary, we have successfully developed the In(III)-pybox complex catalyzed highly enantioselective Mukaiyama aldol reactions between polymeric or hydrated glyoxylate esters and enolsilanes derived from aryl ketones. The enantioenriched β-hydroxy ketones bearing an additional ester group could be obtained in excellent yields and with excellent enantioselectivities under mild conditions. This work not only complements the well-developed Mukaiyama aldol reactions of α-ketone esters but also demonstrates the advantage of In(III)-pybox complexes over other chiral Lewis acids. In addition, solvent and the polymeric or hydrated glyoxylates were used without distillation and the reaction could be carried out in open air, both of which offer great synthetic advantages.
Acknowledgements
We gratefully acknowledge the Nanyang Technological University and Biomedical Research Council (A*STAR Grant No 05/1/22/19/408) for the funding support of this research.Notes and references
- (a) Modern Aldol reactions. Vol. 1: Enolates, Organocatalysis, Biocatalysis and Natural Product Synthesis, ed. R. Mahrwald, Wiley-VCH, Weinheim, 2004 Search PubMed; (b) Modern Aldol reactions. Vol. 2: Metal Catalysis, ed. R. Mahrwald, Wiley-VCH, Weinheim, 2004 Search PubMed; (c) G. Casiraghi, F. Zanardi and G. Appendino, Chem. Rev., 2000, 100, 1929–1972 CrossRef CAS.
- For reviews of asymmetric Mukaiyama aldol reaction, see: (a) T. Bach, Angew. Chem., Int. Ed. Engl., 1994, 33, 417–419 CrossRef; (b) T. K. Hollis and B. Bosnich, J. Am. Chem. Soc., 1995, 117, 4570–4581 CrossRef CAS; (c) H. Groger, E. M. Vogl and M. Shibasaki, Chem.–Eur. J., 1998, 4, 1137–1141 CrossRef CAS; (d) S. G. Nelson, Tetrahedron: Asymmetry, 1998, 9, 357–389 CrossRef CAS; (e) C. Palomo, M. Oiarbide and J. M. Garcia, Chem.–Eur. J., 2002, 8, 36–44 CrossRef CAS; (f) E. M. Carreira, in Comprehensive Asymmetric Catalysis, ed. E. N. Jacobsen, A. Pfaltz and H. Yamamoto, Springer, Berlin, 1999, vol. 1, pp. 997–1065 Search PubMed. For leading literature, see: (g) T. Mukaiyama, K. Narasaka and K. Banno, Chem. Lett., 1973, 1012–1015; (h) K. Saigo, M. Osaki and T. Mukaiyama, Chem. Lett., 1975, 989–990 CAS; (i) T. Mukaiyama, S. Kobayashi, H. Uchiro and I. Shiina, Chem. Lett., 1990, 129–132 CAS.
- For reviews of the direct catalytic asymmetric aldol reaction, see: (a) B. M. Trost and C. S. Brindle, Chem. Soc. Rev., 2010, 39, 1600–1632 RSC; (b) S. Mukherjee, J. W. Yang, S. Hoffmann and B. List, Chem. Rev., 2007, 107, 5471–5569 CrossRef CAS; (c) B. List, Acc. Chem. Res., 2004, 37, 548–557 CrossRef CAS; (d) S. Saito and H. Yamamoto, Acc. Chem. Res., 2004, 37, 570–579 CrossRef CAS; (e) W. Notz, F. Tanaka and C. F. Barbas III, Acc. Chem. Res., 2004, 37, 580–591 CrossRef CAS.
- (a) H. Torii, M. Nakadai, K. Ishihara, S. Saito and H. Yamamoto, Angew. Chem., Int. Ed., 2004, 43, 1983–1986 CrossRef CAS; (b) K. Mei, S. Zhang, S. He, P. Li, M. Jin, F. Xue, G. Luo, H. Zhang, L. Song, W. Duan and W. Wang, Tetrahedron Lett., 2008, 49, 2681–2684 CrossRef CAS; (c) while our manuscript under review, Blanchet and co-workers reported a H8-BINOL-based Brønsted acid catalyzed direct aldol reaction between aryl ketones and glyoxylates for which only moderate enantioselectivities were observed: G. Pousse, F. L. Cavelier, L. Humphreys, J. Rouden and J. Blanchet, Org. Lett., 2010, 12, 3582–3585 Search PubMed.
- (a) B. M. Trost and H. Ito, J. Am. Chem. Soc., 2000, 122, 12003–12004 CrossRef CAS; (b) B. M. Trost, H. Ito and E. R. Silcoff, J. Am. Chem. Soc., 2001, 123, 3367–3368 CrossRef CAS; (c) B. M. Trost and V. S. C. Yeh, Org. Lett., 2002, 4, 3513–3516 CrossRef CAS; (d) H. Li, C. S. Dao, Y. H. Xiao, X. Li and Y. N. Su, J. Org. Chem., 2008, 73, 7398–7401 CrossRef CAS.
- N. Yoshikawa, N. Kumagai, S. Mutsunaga, G. Moll, T. Ohshima, T. Suzuki and M. Shibasaki, J. Am. Chem. Soc., 2001, 123, 2466–2467 CrossRef CAS.
- For recent Mukaiyama-type aldol reactions on aryl ketones with aryl aldehydes, see: (a) S. E. Denmark and J. R. Heemstra, Org. Lett., 2003, 5, 2303–2306 CrossRef CAS; (b) L. Jankowska and J. Mlynarski, J. Org. Chem., 2006, 71, 1317–1321 CrossRef CAS; (c) H. J. Li, H. Y. Tian, Y. J. Chen, L. Liu, D. Wang and C. J. Li, Adv. Synth. Catal., 2005, 347, 1247–1256 CrossRef CAS; (d) K. Ishihara, L. Kondo and H. Yamamoto, J. Org. Chem., 2000, 65, 9125–9128 CrossRef CAS; (e) S. Kiyooka, Y. Takeshita, Y. Tanaka, T. Higaki and Y. Wada, Tetrahedron Lett., 2006, 47, 4453–4456 CrossRef CAS. Other Lewis-acid catalyzed Mukaiyama involving aryl enol silanes: (f) J. Jankowska and J. Mlynarski, J. Org. Chem., 2006, 71, 1317–1321 CrossRef CAS; (g) M. Oishi, S. Aratake and H. Yamamoto, J. Am. Chem. Soc., 1998, 120, 8271–8272 CrossRef CAS; (h) S. Ishikawa, T. Hamada, K. Manabe and S. Kobayashi, J. Am. Chem. Soc., 2004, 126, 12236–12237 CrossRef CAS; (i) M. Sodeoka, K. Ohrai and M. Shibasaki, J. Org. Chem., 1995, 60, 2648–2649 CrossRef CAS; (j) M. Sodeoka, R. Tokunoh, F. Miyazaki, E. Hagiwara and M. Shibasaki, Synlett, 1997, 463–466 CAS.
- (a) M. A. Schwartz and H. E. Van Wart, Prog. Med. Chem., 1992, 29, 271–334 CAS; (b) R. E. Babine and S. L. Bender, Chem. Rev., 1997, 97, 1359–1472 CrossRef; (c) G. M. Coppola and H. F. Schuster, α-Hydroxy Acids in Enantio-selective Synthesis, VCH, Weinheim, Germany, 1997 Search PubMed; (d) A. H. Davidson, A. H. Drummond, W. A. Galloway and M. Whittaker, Chem. Ind., 1997, 3, 258–261; (e) D. E. Levy, F. Lapierre, W. Liang, W. Ye, C. W. Lange, X. Li, D. Grobelny, M. Casabonne, D. Tyrell, K. Holme, A. Nadzan and R. E. Galardy, J. Med. Chem., 1998, 41, 199–223 CrossRef CAS , and references therein.
- For chiral Lewis-acid catalyzed direct asymmetric aldol reactions of keto esters, see: (a) L. C. Akullian, M. L. Snapper and A. H. Hoveyda, J. Am. Chem. Soc., 2006, 128, 6532–6533 CrossRef CAS; (b) D. A. Evans, M. C. Kozlowski, C. S. Burgey and D. W. C. Macmillan, J. Am. Chem. Soc., 1997, 119, 7893–7894 CrossRef CAS; (c) D. A. Evans, C. S. Burgey, M. C. Kozlowski and S. W. Tregay, J. Am. Chem. Soc., 1999, 121, 686–699 CrossRef CAS; (d) M. Langner and C. Bolm, Angew. Chem., Int. Ed., 2004, 43, 5984–5987 CrossRef CAS; (e) P. Remy, M. Langer and C. Bolm, Org. Lett., 2006, 8, 1209–1211 CrossRef CAS; (f) J. Sedelmeier, T. Hammerer and C. Bolm, Org. Lett., 2008, 10, 917–920 CrossRef CAS; (g) M. Langer, P. Remy and C. Bolm, Chem.–Eur. J., 2005, 11, 6254–6265 CrossRef CAS; (h) G. Desimoni, G. Faita, F. Piccinini and M. Toscanini, Eur. J. Org. Chem., 2006, 5228–5230 CrossRef CAS. For organocatalyzed direct asymmetric aldol reactions of keto esters, see: (i) Z. Tang, L. F. Cun, X. Cui, A. Q. Mi, Y. Z. Jiang and L. Z. Gong, Org. Lett., 2006, 8, 1263–1266 CrossRef CAS; (j) X. Y. Xu, Z. Tang, Y. Z. Wang, S. W. Luo, L. F. Cun and L. Z. Gong, J. Org. Chem., 2007, 72, 9905–9913 CrossRef CAS; (k) O. Tokuda, T. Kano, W. G. Gao, T. Ikemoto and K. Maaruoka, Org. Lett., 2005, 7, 5103–5105 CrossRef CAS; (l) J. T. Suri, S. Mitsumori, K. Albertshofe, F. Tanaka and C. F. Barbas III, J. Org. Chem., 2006, 71, 3822–3828 CrossRef CAS; (m) A. Cardova, W. Zou, P. Daiedzic, I. Ibrahem, E. Reyes and Y. Xu, Chem.–Eur. J., 2006, 12, 5383–5397 CrossRef CAS.
- For direct aldol involving glyoxylate, see: (a) R. Dodda and C. G. Zhao, Synlett, 2007, 1605–1609 CAS; (b) M. Markert, U. Scheffler and R. Mahrwald, J. Am. Chem. Soc., 2009, 131, 16642–16643 CrossRef CAS; (c) T. Kano, Y. Yamaguchi, Y. Tanaka and K. Maruoka, Angew. Chem., Int. Ed., 2007, 46, 1738–1740 CrossRef CAS; (d) V. B. Gondi, M. Gravel and V. H. Rawal, Org. Lett., 2005, 7, 5657–5660 CrossRef CAS; (e) T. Urushima, Y. Yasui, H. Ishikawa and Y. Hayashi, Org. Lett., 2010, 12, 2966–2969 CrossRef CAS. For Mukaiyama aldol involving glyoxylate, see: (f) D. A. Evans, D. W. C. MacMillan and K. R. Campos, J. Am. Chem. Soc., 1997, 119, 10859–10860 CrossRef CAS; (g) K. Mikami and S. J. Matsukawa, J. Am. Chem. Soc., 1994, 116, 4077–4078 CrossRef CAS; (h) D. A. Evans, C. E. Masse and J. Wu, Org. Lett., 2002, 4, 3375–3378 CrossRef CAS; (i) D. A. Evans, J. Wu, C. E. Masse and D. W. C. MacMillan, Org. Lett., 2002, 4, 3379–3382 CrossRef CAS.
- (a) D. A. Evans, C. E. Masse and J. Wu, Org. Lett., 2002, 4, 3375–3378 CrossRef CAS; (b) D. A. Evans, J. Wu, C. E. Masse and D. W. C. MacMillan, Org. Lett., 2002, 4, 3379–3382 CrossRef CAS.
- 11% ee was obtained for enolsilane derived from unsubstituted acetophenone in Evans' case.
- (a) N. W. Fadnavis and K. R. Radhika, Tetrahedron: Asymmetry, 2004, 15, 3443–3447 CrossRef CAS; (b) P. Herold, A. F. Indolese, M. Studer, H. P. Jalett, U. Siegrist and H. U. Blaser, Tetrahedron, 2000, 56, 6497–6499 CrossRef CAS; (c) H. U. Blaser, S. Burkhardt, H. J. Kirner, T. Mössner and M. Studer, Synthesis, 2003, 1679–1682 CrossRef CAS.
- R. A. Sheldon, H. J. M. Zeegers, J. P. M. Houbiers and L. A. Hulshof, Chim. Oggi, 1991, 35–47 Search PubMed.
- (a) R. Matsubara, Y. Nakamura and S. Kobayashi, Angew. Chem., Int. Ed., 2004, 43, 3258–3260 CrossRef CAS; (b) M. Terada, K. Soga and N. Momiyama, Angew. Chem., Int. Ed., 2008, 47, 4122–4125 CrossRef CAS.
- For the application of chiral indium complexes in asymmetric synthesis, see: (a) R. Takita, K. Yakura, T. Ohshima and M. Shibasaki, J. Am. Chem. Soc., 2005, 127, 13760–13761 CrossRef CAS; (b) J. Lu, M. L. Hong, S. J. Ji and T. P. Loh, Chem. Commun., 2005, 1010–1012 RSC; (c) J. Lu, M. L. Hong, S. J. Ji, Y. C. Teo and T. P. Loh, Chem. Commun., 2005, 4217–4218 RSC; (d) J. Lu, S. J. Ji, Y. C. Teo and T. P. Loh, Org. Lett., 2005, 7, 159–161 CrossRef CAS; (e) Y. C. Teo, K. T. Tan and T. P. Loh, Chem. Commun., 2005, 1318–1320 RSC; (f) F. Fu, Y. C. Teo and T. P. Loh, Tetrahedron Lett., 2006, 47, 4267–4269 CrossRef CAS; (g) Y. C. Teo and T. P. Loh, Org. Lett., 2005, 7, 2539–2541 CrossRef CAS; (h) Y. C. Teo, J. D. Goh and T. P. Loh, Org. Lett., 2005, 7, 2743–2745 CrossRef CAS; (i) Z. P. Yu, X. H. Liu, Z. H. Dong, M. S. Xie and X. M. Feng, Angew. Chem., Int. Ed., 2008, 47, 1308–1311 CrossRef CAS; (j) X. Zhang, D. H. Chen, X. H. Liu and X. M. Feng, J. Org. Chem., 2007, 72, 5227–5233 CrossRef CAS; (k) N. V. Hanhan, A. H. Sahin, T. W. Chang, J. C. Fettinger and A. K. Franz, Angew. Chem., Int. Ed., 2010, 49, 744–747 CAS.
- (a) J. F. Zhao, H. Y. Tsui, P. J. Wu and T. P. Loh, J. Am. Chem. Soc., 2008, 130, 16492–16493 CrossRef CAS; (b) J. F. Zhao, T. B. Tjan, B. H. Tan and T. P. Loh, Org. Lett., 2009, 11, 5714–5716 CrossRef CAS; (c) J. F. Zhao, B. H. Tan, M. K. Zhu, T. B. Tjan and T. P. Loh, Adv. Synth. Catal., 2010, 352, 2085–2088 CrossRef CAS; (d) J. F. Zhao, T. B. Tjan and T. P. Loh, Tetrahedron Lett., 2010, 51, 5649, DOI:10.1016/j.tetlet.2010.06.066.
- For the effect of 4 Å molecular sieves, see Table 7 of ESI†.
- K. Tran, P. J. Lombardi and J. L. Leighton, Org. Lett., 2008, 10, 3165–3167 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Detailed experimental procedures. See DOI: 10.1039/c0sc00454e |
| ‡ General procedure for the In(III)-pybox complex catalyzed asymmetric Mukaiyama Aldol reactions between polymeric or hydrated glyoxylate esters and aryl ketone-derived enolsilanes: In a 5 mL round-bottom flask containing CH3CN (1.5 ml) with a stirring bar, InBr3 (8.9 mg, 0.025 mmol), pybox (+)-1 (11.8 mg, 0.03 mmol) and 4 Å molecular sieves (150.0 mg) were added and stirred at room temperature for 30 min. To the above mixture, silver hexafluoroantimonate (AgSbF6) (8.6 mg, 0.025 mmol) was added in one portion and stirred for another 30 min. To the pre-prepared catalyst in CH3CN, the polymeric or hydrated glyoxylate ester (1.0 mmol, 2 eq.) was added using a syringe sequentially. The resulting mixture was cooled to −20 °C before enolsilane (0.5 mmol, 1 eq.) was added using syringe under N2 atmosphere. It was stirred until the enolsilane had undergone complete reaction using TLC to monitor its progress. The reaction was quenched by saturated NaHCO3 solution (5 mL). The above mixture was extracted by ethyl acetate three times. The combined organic layer was washed with brine and dried over MgSO4, filtered, and concentrated under reduced pressure. The crude product was loaded directly onto a silica gel column and purified by flash column chromatography to obtain the enantio-enriched β-hydroxy ketones. |
| This journal is © The Royal Society of Chemistry 2011 |