The synthesis and exchange chemistry of frustrated Lewis pair–nitrous oxide complexes†
Rebecca C.
Neu
,
Edwin
Otten
,
Alan
Lough
and
Douglas W.
Stephan
*
Department of Chemistry, University of Toronto, 80 St. George Street, Toronto, Ontario, Canada M5S 3H6. E-mail: dstephan@chem.utoronto.ca
First published on 13th September 2010
Abstract
Facile activation of nitrous oxide is achieved using a series of fluoroarylboranes, B(C6F5)3, PhB(C6F5)2, MesB(C6F5)2, (C6F5)2BOC6F5, B(C6F4-p-H)3, B(C6H4-p-F)3 and 1,4-(C6F5)2BC6F4B(C6F5)2 in the presence of the basic, bulky phosphinetBu3P. Room temperature reaction yields mono- and bis-zwitterionic species of the form tBu3P(N2O)B(C6F5)2R (R = C6F51, Ph 2, Mes 3, OC6F54), tBu3P(N2O)BR3 (R = C6F4-p-H 5, C6H4-p-F 6) and tBu3P(N2O)B(C6F5)2C6F4(C6F5)2B(ON2)PtBu37. N2O activation is similarly achieved using Cy3P and B(C6F4-p-H)3 yielding the zwitterionic species Cy3P(N2O)B(C6F4-p-H)38. Reaction of 6 with [Ph3C][B(C6F5)4] results in facile transfer of the robust tBu3P(N2O) fragment to the stronger Lewis acid Ph3C+ generating [tBu3P(N2O)CPh3][B(C6F5)4] 10. Similarly exchange reactions with titanocene and zirconocene cations generate transition metal and phosphine stabilized nitrous oxide salts, of the form [tBu3P(N2O)MCp2Me][MeB(C6F5)3], (M = Zr 11, Ti 12). The alkoxy zirconocene cation [Cp*2Zr(OMe)]+ forms an FLP in the presence of tBu3P which reacts with N2O providing a direct synthetic route to the corresponding salt [tBu3P(N2O)ZrCp*2(OMe)][B(C6F5)4] 13. Kinetic studies of the self-exchange reaction of tBu3P(N2O)B(C6H4-p-F)3 with B(C6H4-p-F)3 were carried out acquiring information regarding the mechanism of exchange.
Introduction
The issues of ozone depletion and climate change have garnered a considerable amount of attention over the past century due to their apparent negative impact on the environment.1 Anthropogenic emissions of potent greenhouse gases, such as carbon dioxide (CO2), methane (CH4) and nitrous oxide (N2O), continue to rise and contribute to the phenomenon of global warming and to the destruction of the stratospheric ozone layer.1,2 Considerable attention has focused on CO2 and its contribution to global warming, while nitrous oxide (N2O) has remained less recognized despite being roughly 300 times more potent a greenhouse gas than CO2.2N2O is inherently stable and can persist for upwards of 150 years in the stratosphere.3 Recent reports indicate that levels of N2O have risen steadily since industrialization from roughly 270 to 319 ppb, as of 2005, and continue to rise annually.4 In fact, anthropogenic N2O has recently been identified as the largest global ozone depleting gas emission.1In light of the effects of this greenhouse gas, methods to sequester and subsequently break down N2O into less harmful components, such as N2 and H2O, are desirable. In nature, N2O is transformed by metalloenzymes bearing an active site composed of a tetranuclear copper cluster interconnected by a sulfide ligand (Fig. 1).5 The metalloenzyme nitrous oxide reductase is an essential component of the microbial denitrification process, which is a key component of the nitrogen cycle.3,5a,6 This enzyme in bacteria is responsible for catalyzing the transformation of N2O to N2 and H2O and is key to the destruction of N2O produced by the agricultural sector.4
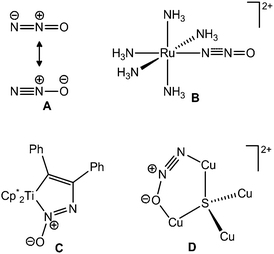 | ||
| Fig. 1 Resonance structures of N2O (A), Taube's Ru–N2O complex (B),8 Hillhouse's N2O insertion product into Cp*2Ti(C2Ph2) (C),10 and the Cu4S active site in nitrous oxide reductase (D).11 | ||
In synthetic chemistry, N2O has been shown to be a strong oxidant yet also a kinetically stable species.7 In coordination chemistry, N2O has been identified as an inherently poor ligand due to its inability to act as either a good σ-donor or π-acceptor (the most important resonance structures are shown in Fig. 1).7 The first example of a transition metal bound N2O complex, [Ru(NH3)5N2O]+, was proposed by Armor and Taube in 1969 (Fig. 1).8 Although this species was never crystallographically confirmed, a number of both computational and spectroscopic studies have since supported this formulation.9 Interestingly, few transition metal–N2O species have been reported in the literature since Armor and Taube's discovery in the late sixties,7 and only recently has an example of a species that contains N2O bound between a phosphine and a Zn center been crystallographically characterized.12N2O is known to undergo reactions at transition metal centers including oxygen insertion into metal–carbon (Fig. 1) and metal–hydride bonds with concomitant N2 liberation10,13 and the oxidation of low-valent metal centers.14 Alternatively N–N scission can be achieved in the presence of metal centers15 while hydrogenation of N2O yields N2 and H2O.16 Very recently, nickel(II) carbene complexes have been shown to react with N2Ovia1,3-dipolar cycloaddition to result in C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond formation with liberation of N2.17 While it is suspected that these reactions proceed through an initial N2O–metal adduct, such claims are yet to be unequivocally substantiated.
O bond formation with liberation of N2.17 While it is suspected that these reactions proceed through an initial N2O–metal adduct, such claims are yet to be unequivocally substantiated.
Recent investigations of the interaction of bulky phosphines with boranes have given rise to the concept of frustrated Lewis pairs (FLPs).18 These systems encompass sterically hindered acids and bases that are precluded from classical Lewis acid–base adduct formation. The resulting unquenched acidity and basicity offer the possibility for further reactivity with a variety of small molecules, such as H2, olefins, dienes, alkynes and carbon dioxide. Such activations have been recently reviewed.18c Herein, the interaction of FLPs with N2O is shown to result in binding of the N2O molecule between the Lewis acidic and basic components in a P–N–N–O–B mode. In addition, the use of Lewis acid exchange chemistry is explored as a route to new N2O compounds. The initial results of this work were communicated previously.19
Results and discussion
Complexation of N2O by FLPs
Treatment of frustrated Lewis pairs consisting of an equimolar mixture of the sterically demanding Lewis base tBu3P and the Lewis acidic boranes R2BR′ with an atmosphere of N2O in a bromobenzene solution resulted in the formation of novel zwitterionic species over a 12 h period at ambient temperatures (Scheme 1).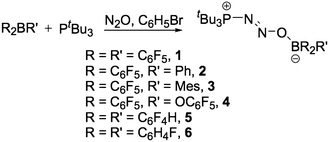 | ||
| Scheme 1 Synthetic method for the generation of 1–6. | ||
Precipitation with hexanes or pentane afforded the zwitterionic species tBu3P(N2O)BR2R′ (R = R′ = C6F5 (1); R = C6F5, R′ = Ph (2); R = C6F5, R′ = Mes (3); R = C6F5, R′ = OC6F5 (4); R = R′ = C6F4-p-H (5); R = R′ = C6H4-p-F (6)) as analytically pure white microcrystalline solids in yields ranging from 76–91%. In an analogous manner, reaction of the linked bisborane (C6F5)2B(C6F4)B(C6F5)2 with two equivalents of tBu3P under an atmosphere of N2O gave the bis-zwitterionic compound tBu3P(N2O)B(C6F5)2C6F4(C6F5)2B(ON2)PtBu3 (7) in good isolated yield (Scheme 2).
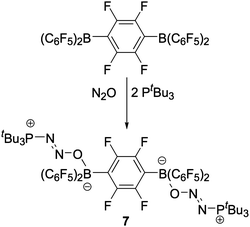 | ||
| Scheme 2 Reaction of the C6F4-linked bisborane with two equivalents of tBu3P and N2O. | ||
Conversely, attempts were made to isolate the corresponding N2O compound from reactions of tBu3P with the weak Lewis acid BPh3 and N2O. Although the desired product was formed in low yield, the white powder isolated upon precipitation with hexanes was ∼90% pure by NMR spectroscopy (Fig. S1) and attempts to further purify the material invariably led to increasing amounts of decomposition. This suggests that a threshold of Lewis acidity is required to generate stable compounds of this type. NMR spectroscopic analyses of compounds 1–7 in CD2Cl2 showed a single resonance in the 31P{1H} NMR spectra between 64 and 69 ppm. 11B{1H} NMR spectra demonstrated a sharp singlet between 0.67 and 6.69 ppm. For species containing C6F5 groups, a significant narrowing of the meta–para gap in comparison to the free boranes was observed in the 19F NMR spectra, with Δ(δ(m-F)–δ(p-F)) around 5 ppm.These data are consistent with 4-coordinate boron centers.20 Compound 4 bears two distinct C6F5 environments (C6F5 and OC6F5) both of which display meta–para gap narrowing upon the generation of the anionic boron center. Upfield 19F NMR shifts are noted for 5 (−134.27/−143.10 ppm) and 6 (−120.87 ppm) relative to the free boranes (−129.60/−138.74 and −106.00 ppm, respectively). The synthesis of the 15N2O isotopologues 1-1515N–7-1515N by reaction of the FLPs with 15N15NO further supported the generation of the P–N–N–O–B species.
The observation of two doublets of doublets in the 15N NMR spectra at roughly 377 and 577 ppm for the terminal and central N atom, respectively, is indicative of two inequivalent and distinct nitrogen environments. 1JN–N coupling constants were found to be approximately 16 Hz, while 1JN–P and 2JN–P were determined to be approximately 60 and 20 Hz, respectively. Comparison of the 15N spectroscopic data of the bound N2O fragment with free N2O (135 and 218 ppm, 1JN–N = 8.1 Hz) indicates that there is a significant perturbation of the N2O fragment upon complexation by the borane and phosphine moieties (Fig. 2). A comparison of the 15N NMR shifts and N–N/N–P coupling constants for compounds 1–7 (Table 1) shows no significant difference upon the reduction of the Lewis acidity of the borane fragment.
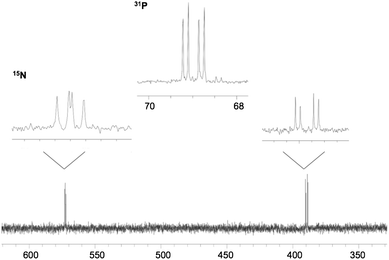 | ||
| Fig. 2 31P{1H} and 15N NMR spectra for 4-1515N. | ||
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 10 | 11 | 13 | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| δ N(1) (ppm) | 381.7 | 377.0 | 375.3 | 389.4 | 367.6 | 381.3 | 382.3 | 376.1 | 412.3 | 398.5 | 386.3 |
| δ N(2) (ppm) | 566.6 | 577.7 | 574.2 | 572.5 | 588.8 | 571.0 | 572.1 | 573.0 | 548.4 | 587.6 | 591.0 |
| 1 J N–N (Hz) | 15.6 | 16.0 | 15.1 | 15.8 | 16.4 | 15.2 | 15.5 | 15.6 | 14.7 | 17.1 | 17.4 |
| 1 J P–N (Hz) | 58.7 | 59.5 | 59.1 | 58.6 | 59.7 | 58.8 | 58.8 | 52.5 | 58.2 | 60.6 | 61.6 |
| 2 J P–N (Hz) | 19.6 | 19.1 | 19.6 | 19.7 | 19.1 | 19.9 | 19.6 | 22.2 | 20.5 | 15.8 | 16.1 |
With the exception of 6, all compounds could be recrystallized by diffusion of pentane or cyclohexane into a CH2Cl2 or C6H5Br solution at room temperature. Crystal structure determinations were carried out for these compounds, unambiguously confirming their formulation, although the quality of the data for compounds 3 and 4 was quite poor and only served to establish connectivities. The molecular structure of 7 is shown in Fig. 3. Comparison of the relevant metrical parameters (Table 2) reveals no significant variation of the bound N2O fragment regardless of the nature of the borane.
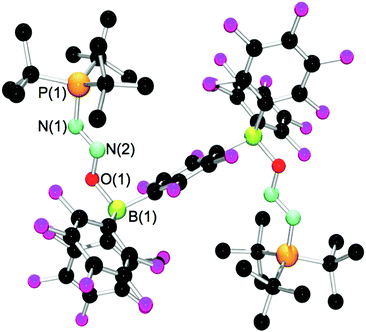 | ||
| Fig. 3 POV-ray depiction of the molecular structure of 7. Hydrogen atoms have been omitted for clarity. | ||
| 1 | 2 | 5 | 7 | 8 | |
|---|---|---|---|---|---|
| a Bond lengths in angstroms (Å), angles in degrees (°). | |||||
| P–N(1) | 1.7087(12) | 1.7107(6) | 1.7092(16) | 1.6905(16) | 1.7146(15) |
| N(1)–N(2) | 1.2573(17) | 1.2602(8) | 1.255(2) | 1.253(2) | 1.260(2) |
| N(2)–O | 1.3363(15) | 1.3270(8) | 1.327(2) | 1.3198(19) | 1.3351(18) |
| O–B | 1.5428(18) | 1.5475(9) | 1.533(2) | 1.536(2) | 1.551(1) |
| P–N(1)–N(2) | 117.04(10) | 112.85(5) | 115.32(13) | 116.48(14) | 111.10(12) |
| N(1)–N(2)–O | 109.11(11) | 111.68(6) | 109.49(14) | 109.81(15) | 111.13(14) |
| N(2)–O–B | 114.43(10) | 111.61(5) | 114.86(14) | 111.26(13) | 110.32(13) |
From the data presented above, it is clear that N2O complexes can be made from combinations of tBu3P with various boron containing Lewis acids demonstrating that a wide range of Lewis acidities is tolerated. It was thus of interest to examine a series of Lewis basic FLP partners for their ability to form analogous N2O complexes. Both phosphorus (Mes3P, (o-tolyl)3P, HPtBu2) and nitrogen bases (2,2,6,6-tetramethylpiperidine, 2,6-lutidine, N-benzylidine-tert-butylamine) were tested in combination with B(C6F5)3, as such FLPs have been shown to effect heterolytic cleavage of H2.18c However, in all cases, treatment with N2O in bromobenzene failed to generate the analogous N2O complexes. Evidently, N2O activation is strongly dependent on the nature of the base suggesting that both strong σ-donation and π-acceptance stabilize the PN2O fragment. Phosphines that are less sterically demanding than tBu3P either form strong adducts with Lewis acids such as B(C6F5)3, or result in nucleophilic aromatic substitution at the para position of a C6F5 ring, as is the case with Cy3P.21 Previously, it has been shown that the borane B(C6F4-p-H)3 is immune to attack at the para-carbon atom, forming an FLP with Cy3P.22 The reaction of an equimolar mixture of Cy3P and B(C6F4-p-H)3 in bromobenzene with an atmosphere of N2O results in formation of Cy3P(N2O)B(C6F4-p-H)38 in 59% isolated yield (Scheme 3). The somewhat lower yield of 8 compared to its tBu3P-analogue 6 is presumably the result of the more facile oxidation of Cy3P by N2O. Indeed, monitoring a solution of Cy3P in C6D5Br under an atmosphere of N2O by 31P NMR showed >90% conversion to Cy3PO overnight, whereas oxidation of tBu3P with N2O is more sluggish (∼50% conversion overnight).23
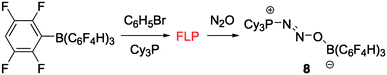 | ||
| Scheme 3 FLP formation from Cy3P and B(C6F4-p-H)3 and subsequent reaction with N2O to give 8. | ||
Spectroscopic analysis of 8 revealed a single resonance in the 31P{1H} NMR spectrum at 56.4 ppm. Two signals, representative of the ortho and meta fluorines, were observed in the 19F NMR spectrum at −134.3 and −143.0 ppm as compared to −130.2 and −139.3 in the free borane.22 A sharp singlet in the 11B{1H} NMR spectrum at 0.8 ppm was observed, typical of a four-coordinate boron center. The isotopically labeled compound 8-1515N shows the expected coupling patterns. Comparison with the 15N spectroscopic data for the tBu3P analogue 5-1515N reveals no significant difference in the 1JN–N coupling constant, whereas 1JP–N is smaller by 7.2 Hz and 2JN–P larger by 2.1 Hz for 8-1515N (Table 1). A crystallographic study unambiguously confirmed the structure of 8 (Fig. 4, pertinent bond lengths and angles in Table 2).
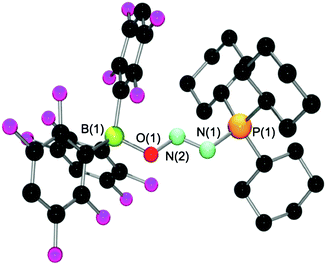 | ||
| Fig. 4 Molecular structure of 8. Hydrogen atoms have been omitted for clarity. | ||
The analogous zwitterionic species using the considerably weaker Lewis acid tris(para-fluorophenyl)borane was sought. An equimolar mixture of B(C6H4-p-F)3 and Cy3P in bromobenzene was allowed to stir overnight under an atmosphere of N2O. Precipitation with pentane yielded a microcrystalline colourless solid. NMR spectroscopy in CD2Cl2 showed singlets in the 11B{1H}, 19F and 31P{1H} NMR spectra at 26.4, −116.4 and 62.6 ppm respectively. Subsequent recrystallization of the crude product from a layered CH2Cl2–pentane solution at −35 °C yielded colorless crystals of Cy3PO(B(C6H4-p-F)3), 9. A crystallographic study confirmed the identity of 9 (Fig. 5). Efforts to observe an intermediate N2O adduct en route to 9 were unsuccessful. The results above are in agreement with the notion that the zwitterionic compounds R3P(N2O)BR′3 are intermediates in Staudinger-type oxidations24 that are kinetically trapped by coordination of the Lewis acidic borane fragment. This kinetic stabilization is only successful when Lewis acid coordination is sufficiently strong (irreversible) and faster than phosphine oxidation. In this sense, these compounds are analogues of the phosphazides R3P(NNN)R′.25
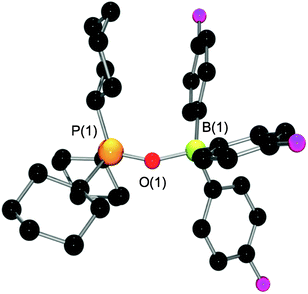 | ||
| Fig. 5 POV-ray depiction of the molecular structure of 9. Hydrogens have been omitted for clarity. Selected bond distances (Å) and angles (°): P(1)–O(1) 1.5251(8); B(1)–O(1) 1.5854(15); P(1)–O(1)–B(1) 147.26(8). | ||
Lewis acid exchange reactions
Further investigation of the use of the weakly acidic borane B(C6H4-p-F)3 for the activation of N2O in conjunction with tBu3P revealed that the borane remains only weakly bound to the tBu3PN2O fragment allowing for facile exchange of the acidic fragment for a stronger Lewis acid.12 Following this method, 1 can easily be generated via the reaction of 6 with an equivalent of B(C6F5)3. Similarly, treatment of 6 with an equivalent of trityl tetrakis(pentafluorophenyl)borate in CD2Cl2 at room temperature immediately yielded a bright yellow solution (Scheme 4).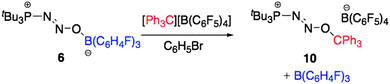 | ||
| Scheme 4 Exchange of the borane fragment B(C6H4-p-F)3 of 6 by trityl cation generating 10. | ||
NMR spectroscopic analysis revealed a new product with a single resonance in the 31P{1H} NMR at 76.3 ppm, considerably downfield from that in 6 (65.4 ppm). NMR spectroscopy indicated the liberation of free B(C6H4-p-F)3, which was easily removed by precipitation of the ionic product and washing with pentane. Synthesis of the 15N isotopologue 10-1515N supported that the tBu3P(N2O) fragment remains intact throughout the exchange process, as evidenced by two doublets of doublets in the 15N NMR spectrum at 548.4 and 412.3 ppm, respectively, with a N–N coupling constant of 14.7 Hz. Similarly the 31P{1H} NMR spectrum revealed a doublet of doublets with 1JP–N and 2JP–N of 58.2 and 20.5 Hz. These data are similar to those observed for 1-1515N to 7-1515N although there is a marked shift for the 15N NMR signals (Table 1). Crystals of 10 were grown from a layered CH2Cl2/cyclohexane solution at room temperature and an X-ray crystallographic study unambiguously confirmed its solid state structure as [tBu3P(N2O)CPh3][B(C6F5)4] (Fig. 6, pertinent bond distances and angles in Table 3).
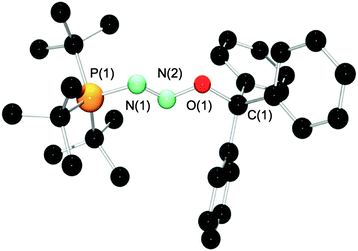 | ||
| Fig. 6 Molecular structure of the cation of 10. Hydrogens and the B(C6F5)4 counter ion have been omitted for clarity. | ||
| 10 (X = C) | 11 (X = Zr) | 13 (X = Zr) | |
|---|---|---|---|
| a Bond lengths in angstroms (Å), angles in degrees (°). | |||
| P(1)–N(1) | 1.7217(13) | 1.719(4) | 1.683(3) |
| N(1)–N(2) | 1.2418(17) | 1.233(5) | 1.266(4) |
| N(2)–O(1) | 1.3752(15) | 1.372(5) | 1.300(4) |
| O(1)–X(1) | 1.4955(17) | 2.105(3) | 2.151(3) |
| P(1)–N(1)–N(2) | 115.98(10) | 111.3(3) | 116.9(3) |
| N(1)–N(2)O(1) | 108.52(12) | 111.0(3) | 111.9(3) |
| N(2)–O(1)X(1) | 110.68(10) | 126.2(2) | 123.4(2) |
Throughout the exchange process the tBu3P(N2O) fragment remains intact and appears to be inherently stable. It is worth noting that this method of borane exchange also offers an easy and clean route to the generation of 2, 3, 4, 5 and 7via exchange of 6 with one equivalent of PhB(C6F5)2, MesB(C6F5)2, (C6F5)2BOC6F5, B(C6F4-p-H)3 or with half an equivalent of 1,4-(C6F5)2B(C6F4)B(C6F5)2.
Extending this borane exchange strategy to early transition metal complexes is useful as there are few documented cases where intact N2O interacts with metal complexes without the decomposition of N2O to O and N2.10 Thus, treatment of a bromobenzene solution of [Cp2ZrMe][MeB(C6F5)3] with an equivalent of 6 resulted in a beige oil that solidified upon trituration with pentane. Recrystallization from CH2Cl2–pentane at −35 °C afforded 11 in 83% isolated yield (Scheme 5). NMR spectroscopic studies revealed a singlet in the 31P{1H} NMR at 67.7 ppm. Three peaks were observed in the 19F NMR spectrum pertaining to the ortho, para and meta signals of the MeB(C6F5)3 anion at −133.1, −165.3 and −167.8 ppm, with no traces of residual B(C6H4-p-F)3. A broad signal in the 1H NMR at 0.48 ppm was observed pertaining to the methyl group of the counter anion, MeB(C6F5)3. With these spectroscopic data, [tBu3P(N2O)ZrCp2Me][MeB(C6F5)3] was proposed as the structure of 11, which was confirmed by a single crystal X-ray diffraction study (Fig. 7, Table 3).
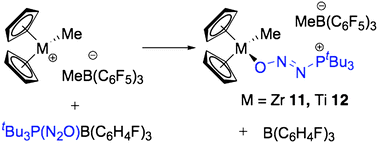 | ||
| Scheme 5 Synthetic scheme for the generation of 11 and 12 by Lewis acid exchange. | ||
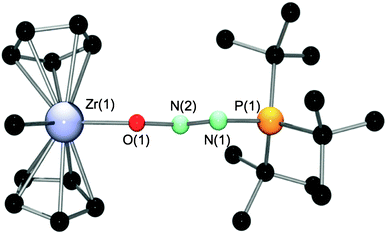 | ||
| Fig. 7 The molecular structure of the cation of 11. Hydrogens and the counter anion MeB(C6F5)3 have been omitted for clarity. | ||
The crystallographic characterization of 11 constitutes the only example of an intact N2O fragment where the oxygen of the N2O fragment is bound directly to a zirconium center.10 As observed in the tBu3P(N2O)BR3 compounds described above, the molecular structure displays the same trans orientation of the ZrO and tBu3P fragments relative to the N–N bond. The metric parameters for the PN2O fragment in 11 are similar to those observed in the borane compounds described above. The N(2)–O(1)–Zr(1) angle of 126.2(2)° is relatively large in comparison to the corresponding N–O–B or N–O–C angles in 1–10 (110.32(13)–114.86(14)°), which may reflect some degree of interaction between an O lone pair and a vacant metal-based orbital that is absent in the main group species 1–10. Coordination of the tBu3P(N2O) fragment to the cationic zirconocene center results in little structural changes in the Cp2ZrMe+ moiety. The Cp(centroid)–Zr–Cp(centroid) angle in 11 is 130.5°, which may be compared to 129.6 in [Cp2ZrMe(THF)][BPh4]26 or 129.2 in Cp2ZrCl2.27 As well, the Zr–Me (2.261(4) Å) and Zr–O (2.105(3) Å) bond lengths are statistically indistinguishable from the corresponding distances in Cp2ZrMe(THF)+ (2.256(10) and 2.122(14) Å, respectively).26 In a similar manner, Lewis acid exchange between [Cp2TiMe][MeB(C6F5)3] and 6 resulted in the formation of [tBu3P(N2O)TiCp2Me][MeB(C6F5)3] 12, in 81% yield. These species were found to be stable in solution for a number of days.
It is noteworthy that attempts to generate 11 directly from the reaction of the FLP generated by equimolar mixture of [Cp2ZrMe][MeB(C6F5)3] and with 15N2O gave only inseparable and unidentified reaction products. However, in contrast, an analogous species, generated by the zirconocene methoxide cation [Cp*2Zr(OMe)][B(C6F5)4] and an equivalent of tBu3P in bromobenzene (Scheme 6) reacted under an atmosphere of N2O for 12 h, to give a yellow solid, 13, which was isolated in 74% yield (Scheme 6). Spectroscopic analysis revealed a single resonance in the 31P{1H} NMR at 64.4 ppm while the 1H NMR data showed resonances attributable to the Cp* and methoxide ligands at 1.91 and 3.98 ppm, respectively. The 11B{1H} and 19F NMR spectra were in keeping with the corresponding B(C6F5)4 counter anion. In addition, 15N and 31P NMR spectroscopy on the isotopically labeled compound 13-1515N is indicative of an intact tBu3P(N2O) fragment (Table 1). The sum of these data firmly supports the formulation of 13 as [tBu3P(N2O)ZrCp*2(OMe)][B(C6F5)4].
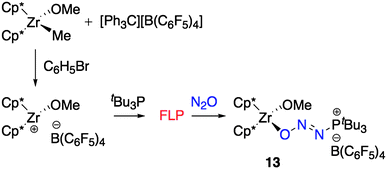 | ||
| Scheme 6 Activation of N2O employing a Zr/P FLP yielding 13. Cp* = C5Me5. | ||
An X-ray structure determination of 13 (Fig. 8, pertinent bond lengths and angles in Table 3) confirmed that the N2O fragment is O-bound to the Zr center and N-bound to the P center of the phosphine while the orientation remains trans about the N–N bond. Based on the observation of quite disparate Zr–O bond distances, the OMe and ON2tBu3P moieties interact with the metal center in a rather different manner. The Zr–O(alkoxide) bond length in 13 of 1.916(2) Å is typical for zirconocene alkoxides (e.g., [Cp2Zr(OtBu)(THF)][BPh4]: average 1.890(4) Å)28 but significantly shorter than the Zr–ON2tBu3P distance of 2.151(3) Å. In addition, the large Zr(1)–O(2)–C(21) angle of 169.9(2)° in comparison to Zr(1)–O(1)–N(2) (123.4(2)°) suggests that the methoxide ligand, but not the ON2tBu3P fragment, is engaged in substantial backdonation to the electron deficient metal center.28,29 A comparison between 11 and 13 shows that the Zr(1)–O(1) bond is shortest in 11, in agreement with it having the most Lewis acidic metal center. The P(1)–N(1) and N(2)–O(1) bond lengths in 13 of 1.683(3) and 1.300(4) Å, respectively, were found to be shorter than those in 11 (1.719(4) and 1.372(5) Å). Conversely, the N(1)–N(2) bond in 13 is somewhat longer (1.266(4) vs. 1.233(5) Å in 11). Unlike the P/B systems where variation of the Lewis acidic fragment has been shown to have very little effect on the nature of the N2O moiety, variation in the Lewis acidity of the metallocene cation causes a more significant perturbation of the PN2O fragment.
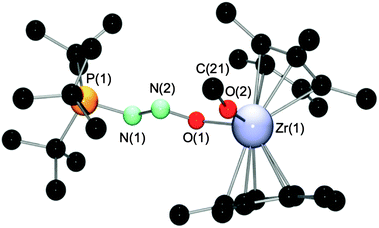 | ||
| Fig. 8 The molecular structure of 13. Hydrogens and the counter anion B(C6F5)4 have been omitted for clarity. | ||
Mechanism of borane exchange
In order to obtain mechanistic insight, a kinetic study of Lewis acid exchange reactions was undertaken. Thus, to a solution of tBu3P(N2O)B(C6H4-p-F)36 in CD2Cl2 at room temperature was added an equimolar amount of a stronger Lewis acid (e.g., B(C6F5)3, B(C6F5)2Ph). The NMR spectra taken immediately after mixing (<5 min) in all cases showed only the presence of exchange products, suggesting that these reactions are fast. It is likely that the weakly Lewis acidic nature of the borane in 6 results in facile exchange. Employing a species containing a more tightly bound Lewis acid, the trityl compound [tBu3P(N2O)CPh3][B(C6F5)4] 10 was shown to react cleanly with B(C6F5)3 to give [Ph3C][B(C6F5)4] and tBu3P(N2O)B(C6F5)3 (1). However, even when CD2Cl2 was condensed at −196 °C into an NMR tube containing solid 10 and B(C6F5)3, NMR spectroscopic analysis of the reaction at −80 °C showed full conversion to the expected products before the first spectrum could be measured. It was thus not possible to obtain rate data. As an alternative, the exchange between tBu3P(N2O)B(C6F5)3 (1) and free B(C6F5)3 was examined by 19F NMR spectroscopy. Well-separated, sharp resonances for both species are observed in the 19F NMR spectrum, but 2D 19F EXSY experiments30 (τmix = 1.2 s) showed no evidence of exchange between the two species up to 80 °C. Gratifyingly, exchange was observed between tBu3P(N2O)B(C6H4-p-F)3 (6) and free B(C6H4-p-F)3 at room temperature and analysis of 2D 19F EXSY spectra obtained between −25 and +25 °C allowed determination of the activation parameters of the exchange process as ΔH‡ = 71.2(9) kJ mol−1 and ΔS‡ = 32(3) J mol−1 K−1 (see Supporting Information for details). The small, positive value for ΔS‡ suggests that the B–O linkage is somewhat weakened before the incoming borane binds. The lack of observable exchange (i.e., a significantly higher activation barrier) between 1, in which the borane is more strongly bound, and B(C6F5)3 is in agreement with this proposal.31Conclusions
FLPs react with N2O under mild conditions to give zwitterionic products R3P(N2O)BR2R′ in high yield. Phosphines with diminished steric demands or Lewis basicity, or nitrogen Lewis bases do not give analogous compounds, suggesting that a combination of strong σ-donation and π-acceptance is required to capture the N2O fragment. A range of borane Lewis acidities have been studied. These show little difference in the geometric or electronic properties of the resulting N2O moieties. The robustness of the R3P(N2O) moiety allows the synthesis of otherwise inaccessible N2O species by exchange of the borane for stronger Lewis acids such as Ph3C+ and [Cp2ZrMe]+. This route to N2O transition metal derivatives offers a new strategy to exploring and understanding the nature of N2O binding, transport and reduction in synthetic as well as biological systems. Further studies of both main group and transition N2O species are underway and will be reported in due course.Acknowledgements
DWS gratefully acknowledges the financial support of NSERC of Canada and the award of a Canada Research Chair and a Killam Research Fellowship. EO is grateful for the support of a Rubicon postdoctoral fellowship from the Netherlands Organisation for Scientific Research (NWO). RCN is grateful for the award of an Ontario Graduate Scholarship.Notes and references
- A. R. Ravishankara, J. S. Daniel and R. W. Portmann, Science, 2009, 326, 123–125 CrossRef CAS.
- J. Hansen and M. Sato, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 16109–16114 CrossRef CAS.
- D. Richardson, H. Felgate, N. Watmough, A. Thomson and E. Baggs, Trends Biotechnol., 2009, 27, 388–397 CrossRef CAS.
- C. S. Snyder, T. W. Bruulsema, T. L. Jensen and P. E. Fixen, Agric. Ecosyst. Environ., 2009, 133, 247–266 CrossRef CAS.
- (a) W. G. Zumft and P. M. H. Kroneck, in Advances in Microbial Physiology, ed. K. P. Robert, Academic Press, 2006, pp. 107–227 Search PubMed; (b) E. I. Solomon, R. Sarangi, J. S. Woertink, A. J. Augustine, J. Yoon and S. Ghosh, Acc. Chem. Res., 2007, 40, 581–591 CrossRef CAS.
- W. C. Trogler, J. Chem. Educ., 1995, 72, 973 CrossRef CAS.
- W. B. Tolman, Angew. Chem., Int. Ed., 2010, 49, 1018–1024 CrossRef CAS.
- J. N. Armor and H. Taube, J. Am. Chem. Soc., 1969, 91, 6874–6876 CrossRef CAS.
- (a) F. Bottomley and J. R. Crawford, J. Am. Chem. Soc., 1972, 94, 9092–9095 CrossRef CAS; (b) F. Bottomley and W. V. F. Brooks, Inorg. Chem., 1977, 16, 501–502 CrossRef CAS; (c) D. F. T. Tuan and R. Hoffmann, Inorg. Chem., 1985, 24, 871–876 CrossRef CAS; (d) F. Paulat, T. Kuschel, C. Nather, V. K. K. Praneeth, O. Sander and N. Lehnert, Inorg. Chem., 2004, 43, 6979–6994 CrossRef CAS.
- G. A. Vaughan, C. D. Sofield, G. L. Hillhouse and A. L. Rheingold, J. Am. Chem. Soc., 1989, 111, 5491–5493 CrossRef CAS.
- P. Chen, S. I. Gorelsky, S. Ghosh and E. I. Solomon, Angew. Chem., Int. Ed., 2004, 43, 4132–4140 CrossRef CAS.
- R. C. Neu, E. Otten and D. W. Stephan, Angew. Chem., Int. Ed., 2009, 48, 9709–9712 CAS.
- (a) G. A. Vaughan, P. B. Rupert and G. L. Hillhouse, J. Am. Chem. Soc., 1987, 109, 5538–5539 CrossRef CAS; (b) A. W. Kaplan and R. G. Bergman, Organometallics, 1997, 16, 1106–1108 CrossRef CAS.
- (a) F. Bottomley, Polyhedron, 1992, 11, 1707–1731 CrossRef CAS; (b) W. A. Howard and G. Parkin, J. Am. Chem. Soc., 1994, 116, 606–615 CrossRef CAS; (c) J. T. Groves and J. S. Roman, J. Am. Chem. Soc., 1995, 117, 5594–5595 CrossRef CAS; (d) H. Z. Yu, G. C. Jia and Z. Lin, Organometallics, 2009, 28, 1158–1164 CrossRef CAS.
- (a) J.-P. F. Cherry, A. R. Johnson, L. M. Baraldo, Y.-C. Tsai, C. C. Cummins, S. V. Kryatov, E. V. Rybak-Akimova, K. B. Capps, C. D. Hoff, C. M. Haar and S. P. Nolan, J. Am. Chem. Soc., 2001, 123, 7271–7286 CrossRef CAS; (b) A. R. Johnson, W. M. Davis, C. C. Cummins, S. Serron, S. P. Nolan, D. G. Musaev and K. Morokuma, J. Am. Chem. Soc., 1998, 120, 2071–2085 CrossRef CAS; (c) C. E. Laplaza, A. L. Odom, W. M. Davis, C. C. Cummins and J. D. Protasiewicz, J. Am. Chem. Soc., 1995, 117, 4999–5000 CrossRef CAS.
- J.-H. Lee, M. Pink, J. Tomaszewski, H. Fan and K. G. Caulton, J. Am. Chem. Soc., 2007, 129, 8706–8707 CrossRef CAS.
- N. D. Harrold, R. Waterman, G. L. Hillhouse and T. R. Cundari, J. Am. Chem. Soc., 2009, 131, 12872–12873 CrossRef CAS.
- (a) D. W. Stephan, Org. Biomol. Chem., 2008, 6, 1535–1539 RSC; (b) D. W. Stephan, Dalton Trans., 2009, 3129–3136 RSC; (c) D. W. Stephan and G. Erker, Angew. Chem., Int. Ed., 2010, 49, 46–76 CAS.
- E. Otten, R. C. Neu and D. W. Stephan, J. Am. Chem. Soc., 2009, 131, 9918–9919 CrossRef CAS.
- W. E. Piers, in Adv. Organomet. Chem., ed. R. West and A. F. Hill, Academic Press, 2004, pp. 1–76 Search PubMed.
- (a) G. C. Welch, L. Cabrera, P. A. Chase, E. Hollink, J. D. Masuda, P. R. Wei and D. W. Stephan, Dalton Trans., 2007, 3407–3414 RSC; (b) G. C. Welch, T. Holtrichter-Roessmann and D. W. Stephan, Inorg. Chem., 2008, 47, 1904–1906 CrossRef CAS.
- M. Ullrich, A. J. Lough and D. W. Stephan, J. Am. Chem. Soc., 2009, 131, 52–53 CrossRef CAS.
- S. Poh, R. Hernandez, M. Inagaki and P. G. Jessop, Org. Lett., 1999, 1, 583–586 CrossRef CAS.
- E. H. H. Staudinger, Helv. Chim. Acta, 1921, 4, 861–886 CrossRef CAS.
- M. W. P. Bebbington and D. Bourissou, Coord. Chem. Rev., 2009, 253, 1248–1261 CrossRef CAS.
- R. F. Jordan, C. S. Bajgur, R. Willett and B. Scott, J. Am. Chem. Soc., 1986, 108, 7410–7411 CrossRef CAS.
- J. Y. Corey, X.-H. Zhu, L. Brammer and N. P. Rath, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1995, 51, 565–567 CrossRef.
- S. Collins, B. E. Koene, R. Ramachandran and N. J. Taylor, Organometallics, 1991, 10, 2092–2094 CrossRef CAS.
- J. W. Lauher and R. Hoffmann, J. Am. Chem. Soc., 1976, 98, 1729–1742 CrossRef CAS.
- (a) C. L. Perrin and T. J. Dwyer, Chem. Rev., 1990, 90, 935–967 CrossRef CAS; (b) P. Espinet, A. C. Albéniz, J. A. Casares and J. M. Martínez-Ilarduya, Coord. Chem. Rev., 2008, 252, 2180–2208 CrossRef CAS.
- An alternative explanation for the positive activation entropy could be a change in the solvation (CH2Cl2) of the zwitterionic compound 6. We were not able to test this hypothesis due to the poor solubility of 6 in non-polar solvents.
Footnote |
| † Electronic supplementary information (ESI) available: Crystallographic data (CIF format) for 1, 2, 5, 7, 8–11 and 13, synthesis and spectroscopic and analytical data for all new compounds. NMR spectra of the product from reaction of tBu3P and BPh3 with N2O, and details for the 19F EXSY NMR analysis. CCDC reference numbers 768908 and 786852–786857. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c0sc00398k |
| This journal is © The Royal Society of Chemistry 2011 |