Catalytic nitrene transfer by a zirconium(IV) redox-active ligand complex†
Andy I.
Nguyen
,
Ryan A.
Zarkesh
,
David C.
Lacy
,
Megan K.
Thorson
and
Alan F.
Heyduk
*
Department of Chemistry, University of California, 1102 Natural Sciences 2, Irvine, California 92697-2025, USA. E-mail: aheyduk@uci.edu
First published on 27th October 2010
Abstract
Nitrene transfer catalyzed by a d0zirconium(IV) complex with a redox-active ligand is reported. The redox-active ligand, bis(2-isopropylamido-4-methoxyphenyl)amide ([NNNcat]3−), afforded zirconium(IV) complexes, [NNNcat]ZrClL2 (1a, L = THF; 1b, L = CNtBu; 1c, L = py), upon reaction with ZrCl4(THF)2. Complex 1a was oxidized by one and two electrons using PhICl2, affording [NNNsq•]ZrCl2(THF) (2) and [NNNq]ZrCl3 (3), respectively. Aryl azides reacted with 1a to afford zirconium imide dimers, including the crystallographically characterized species {[NNNq]ZrCl(μ2-p-NC6H4tBu)}2 (4). The formation of 4 is the result of the addition of an aryl nitrene to the zirconium(IV) metal center. When 1b was reacted with organoazides, the dimer was not observed, but rather the nitrene group was transferred to the isonitrile to form a carbodiimide. In the presence of excess organoazide and isonitrile, catalytic carbodiimide formation occurred, showing that a redox-active ligand and a d0 metal center can work in concert to effect nitrene group transfer reactivity.
Introduction
The reactivity of transition metal imido complexes is highly dependent on the nature of the metal–imido interaction.1,2 Imido complexes of electrophilic early transition metals have highly polarized metal–nitrogen multiple bonds that enable non-redox reactivity such as catalytic hydroamination3–6 and C–H bond activation.7–10 In contrast, imido complexes of late transition metals are less polarized, but can effect olefin aziridination11–13 and C–H bond amination.14–19 It would be attractive to combine the features of early- and late-metal imido complexes such that species both with highly polarized metal–imido bonds and capable of two-electron reactivity could be studied. Towards this end, electrophilic late-transition metal imido complexes have been examined for group-transfer reactivity,20,21 and in a few novel cases, two-electron atom-transfer reactivity has been observed from vanadium nitride complexes.22,23 Another versatile strategy for two-electron group-transfer reactivity is to incorporate redox-active auxiliary ligands into early transition metal imido complexes. This strategy offers many attractive features, including the ability to tune both steric and electronic parameters of the metal–imido complex through modifications to the redox-active ligand.In an effort to bridge the gap in reactivity between early- and late-transition metal imido complexes, we utilize redox-active ligands that are capable of multi-electron valence changes. The incorporation of redox-active ligands on early transition metals has been shown to enable oxidative addition at d0 metals24–27 and reductive elimination to form C–C28 and N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N29 bonds. Recently, we have used the redox-active ligand bis(2-isopropylamido-4-methoxyphenyl)amide ([NNNcat]3−) to enable the transfer of a nitrene from an organic azide to a tantalum(V) metal center (Scheme 1),30 in the process oxidizing the ligand from the catecholate to the quinonate form ([NNNq]−). While the nitrene transfer reaction to tantalum(V) proved the concept that two-electron, group-transfer to a d0 metal could be engendered by a redox-active ligand, the tantalum–imido product proved to be relatively unreactive. With the ultimate goal of developing nitrene transfer catalysts based on electrophilic early transition metals and redox-active ligands, it became imperative to determine if the irreversibility of this nitrene transfer to tantalum was an inherent characteristic of the d0-metal, redox-active-ligand strategy, or if the irreversibility was simply a consequence of the specific coordination environment of the tantalum–imido product. In an attempt to exploit the reactivity of the metal–imido functional group, we sought access to a coordinatively unsaturated analog of the [NNNq]TaCl2(NPh) complex.
N29 bonds. Recently, we have used the redox-active ligand bis(2-isopropylamido-4-methoxyphenyl)amide ([NNNcat]3−) to enable the transfer of a nitrene from an organic azide to a tantalum(V) metal center (Scheme 1),30 in the process oxidizing the ligand from the catecholate to the quinonate form ([NNNq]−). While the nitrene transfer reaction to tantalum(V) proved the concept that two-electron, group-transfer to a d0 metal could be engendered by a redox-active ligand, the tantalum–imido product proved to be relatively unreactive. With the ultimate goal of developing nitrene transfer catalysts based on electrophilic early transition metals and redox-active ligands, it became imperative to determine if the irreversibility of this nitrene transfer to tantalum was an inherent characteristic of the d0-metal, redox-active-ligand strategy, or if the irreversibility was simply a consequence of the specific coordination environment of the tantalum–imido product. In an attempt to exploit the reactivity of the metal–imido functional group, we sought access to a coordinatively unsaturated analog of the [NNNq]TaCl2(NPh) complex.
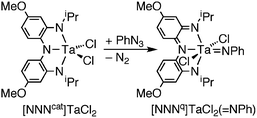 | ||
| Scheme 1 Redox-active ligand-enabled nitrene transfer to TaV. | ||
Results and discussion
Zirconation of the [NNNcat]3− ligand afforded six-coordinate zirconium(IV) complexes. Triple deprotonation of [NNNcat]H3 with nBuLi and subsequent treatment with ZrCl4(THF)2 yielded [NNNcat]ZrCl(THF)2 (1a) as an orange powder in good yields. Complex 1a is a six-coordinate, C2v-symmetric complex as indicated by the presence of only one methyl and one methine resonance for the ligand isopropyl groups in the 1H NMR spectrum. Two equivalent THF ligands observed in the 1H NMR spectrum, along with the absence of any N–H resonances, are consistent with a meridional coordination of the [NNNcat]3− ligand, as observed for its coordination to tantalum(V).30 The symmetry of both the THF resonances and the ligand isopropyl resonances suggest that the chloride ligand sits trans to the diphenylamide nitrogen of the [NNNcat]3− ligand. The UV-vis absorption spectrum shows a ligand-to-metal-charge-transfer (LMCT) band at 306 nm (13![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 700 M−1 cm−1) tailing into the visible region of the spectrum, consistent with the orange color of the complex.
700 M−1 cm−1) tailing into the visible region of the spectrum, consistent with the orange color of the complex.
The THF ligands of 1a were readily exchanged for more strongly donating ligands. tert-Butyl isocyanide reacted immediately with 1a to form [NNNcat]ZrCl(CNtBu)2 (1b) as a deep red–purple solid, while pyridine reacted with 1a to form [NNNcat]ZrCl(py)2 (1c) as a brown solid. The NMR spectra of 1b and 1c are analogous to those for 1a. The solid-state IR spectrum of 1b shows a single C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N stretch at 2201 cm−1, again consistent with a C2v-symmetric molecule. This CN stretch for 1b is shifted to higher frequency than the CN stretch for free tBuNC (2125 cm−1), consistent with the assignment of a [NNNcat]ZrIV complex.
N stretch at 2201 cm−1, again consistent with a C2v-symmetric molecule. This CN stretch for 1b is shifted to higher frequency than the CN stretch for free tBuNC (2125 cm−1), consistent with the assignment of a [NNNcat]ZrIV complex.
To establish the redox-reactivity of the [NNNcat]ZrIV platform, one- and two-electron halogen oxidation reactions of 1a were examined. The redox reactivity of the [NNNcat]ZrClL2 platform is summarized in Scheme 2. Addition of one half of an equivalent of PhICl2 to 1a resulted in a color change from orange to deep wine-red with the formation of the complex [NNNsq•]ZrCl2(THF) (2), containing the one-electron oxidized, semiquinonate form of the redox-active ligand. Complex 2 is a paramagnetic, S = ½ complex and displays a broad isotropic signal in the X-band EPR spectrum at 298 K, consistent with an unpaired electron on an organic ligand (g = 1.999; aN = 14 G; see ESI†). The unpaired electron of 2 gives rise to an absorbance in the visible region of the UV-vis spectrum at 408 nm (1900 M−1 cm−1). When 1a was treated with one equivalent of PhICl2, a forest-green microcrystalline solid was isolated and characterized as [NNNq]ZrCl3 (3), containing the two-electron oxidized, quinonate form of the redox-active ligand. Diamagnetic 3 was characterized by 1H and 13C NMR spectroscopy. Notably, the 1H NMR spectrum shows ca. + 0.5 ppm shifts in the resonances for the OMe and iPr groups, consistent with oxidation of the redox-active ligand to the quinonate state. Similarly, the UV-vis spectrum of 3 shows two strong absorption bands in the visible region at 480 (4200 M−1 cm−1) and 865 (4500 M−1 cm−1) nm, as observed for d0[NNNq]TaV complexes.30
![Reactivity of [NNNcat]ZrClL2 (1a–c, L = THF, CNtBu, py).](/image/article/2011/SC/c0sc00414f/c0sc00414f-s2.gif) | ||
| Scheme 2 Reactivity of [NNNcat]ZrClL2 (1a–c, L = THF, CNtBu, py). | ||
The two-electron oxidation of 1a by halogens indicated that other two-electron additions would be possible at the zirconium center. Orange solutions of 1a reacted with p-tert-butylphenyl azide to release N2 gas with a concomitant color change to olive green. Isolation of the zirconium product afforded the bimetallic bis(imide) complex {[NNNq]ZrCl(μ2-p-NC6H4tBu)}2 (4) as determined by single-crystal X-ray diffraction‡ (see ESI†), 1H and 13C NMR spectroscopy, and solution molecular weight measurements (measured 1074 g mol−1; actual 1229 g mol−1). Reactions between 1a and other aryl azides (PhN3, p-CF3C6H4N3, 2,4,6-Me3C6H2N3) gave analogous dimers that were characterized by NMR spectroscopy.
Dimer 4 is an edge-sharing bioctahedron with the imido ligands bridging the metal centers. Fig. 1 displays the molecular structure of 4 as an ORTEP; selected metrical parameters for the core of the zirconium dimer are given in Table 1. The molecule has two chloride ligands occupying syn coordination sites with respect to the Zr⋯Zr vector, placing both [NNNq]− ligands on the same side of the dimer. This configuration generates inequivalent environments for the two bridging imido ligands, a feature which is preserved in solution as evidenced by two 1H NMR signals at 1.07 and 1.27 ppm for the tBu groups of the bridging imido ligands. Overall, 4 shows C2v symmetry in solution with the rotation axis defined by the bridging imido nitrogen atoms. Bond lengths within the redox-active ligand are consistent with the quinonate oxidation state of the ligand,30 and short C–O distances (average 1.36 Å) provide evidence for donation of the methoxy lone electron pairs into the quinonate π manifold. The Zr⋯Zr distance is long at 3.24 Å and Zr–N(imide) distances are normal for imide ligands bridging two zirconium centers.31–33 Steric crowding within the dimer is evident from the orientation of the ligand isopropyl groups, which are rotated to minimize interactions between the methyl groups across the dimer.
![ORTEP diagram for {[NNNq]ZrCl(μ2-p-NC6H4tBu)}2 (4). Thermal ellipsoids are shown at 50% probability. Hydrogen atoms and a diethyl ether molecule have been omitted for clarity.](/image/article/2011/SC/c0sc00414f/c0sc00414f-f1.gif) | ||
| Fig. 1 ORTEP diagram for {[NNNq]ZrCl(μ2-p-NC6H4tBu)}2 (4). Thermal ellipsoids are shown at 50% probability. Hydrogen atoms and a diethyl ether molecule have been omitted for clarity. | ||
| Zr(1)–Cl(1) | 2.5230(8) | Zr(2)–Cl(2) | 2.5316(8) |
| Zr(1)–N(1) | 2.362(2) | Zr(2)–N(4) | 2.365(2) |
| Zr(1)–N(2) | 2.248(2) | Zr(2)–N(6) | 2.276(2) |
| Zr(1)–N(3) | 2.244(2) | Zr(2)–N(5) | 2.229(2) |
| Zr(1)–N(7) | 2.107(2) | Zr(2)–N(8) | 2.121(2) |
| Zr(1)–N(8) | 2.103(2) | Zr(2)–N(7) | 2.088(2) |
| N(7)–Zr(1)–N(8) | 79.29(9) | N(7)–Zr(2)–N(8) | 79.33(9) |
| N(2)–Zr(1)–N(3) | 137.57(9) | N(5)–Zr(2)–N(6) | 136.36(9) |
| Cl(1)–Zr(1)–N(1) | 93.21(6) | Cl(2)–Zr(2)–N(4) | 100.25(6) |
| Zr(1)–N(7)–Zr(2) | 101.15(9) | Zr(1)–N(8)–Zr(2) | 100.22(9) |
Treatment of isocyanide derivative 1b with organic azides resulted in nitrene transfer to form the corresponding carbodiimide product. Complex 1b reacted with 1 equiv. of p-tert-butylphenyl azide at 25 °C to generate N2 gas and the carbodiimide adduct of the [NNNcat]ZrCl fragment, which was characterized by conversion to 1c. While reactions with aryl azides stopped after the production of two equivalents of carbodiimide, 10 equiv. of AdN3 or tBuN3 reacted with 10 equiv. of tBuNC in the presence of 1b to give nitrene transfer and the catalytic formation of AdNCNtBu or tBuNCNtBu, respectively. The coupling of tBuNC with AdN3 reacted to completion in 2 h at 55 °C with 10 mol% of 1b. Kinetics experiments gave the empirical rate law for carbodiimide formation shown in eqn (1) with kobs = 0.123(7) s−1 at 55 °C. Based on these data a mechanism for carbodiimide formation is proposed in Scheme 3.
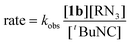 | (1) |
![Mechanism for nitrene transfer catalyzed by [NNNcat]ZrCl(CNtBu)2 (1b).](/image/article/2011/SC/c0sc00414f/c0sc00414f-s3.gif) | ||
| Scheme 3 Mechanism for nitrene transfer catalyzed by [NNNcat]ZrCl(CNtBu)2 (1b). | ||
The mechanism of nitrene transfer presented in Scheme 3 has several interesting features. Dissociation of an isonitrile ligand from coordinatively-saturated 1b to open up a reactive site (step a) is consistent with the inverse dependence of the reaction rate on the concentration of free isonitrile. That this ligand dissociation is fast and reversible is consistent with the exchange reactions used to prepare 1b and 1c from 1a. The second and rate-determining step leading to carbodiimide formation is the transfer of nitrene from the organic azide to the zirconium(IV) center (step b in Scheme 2). To compensate the formation of the zirconium–nitrogen multiple bond, the redox-active ligand is oxidized by two electrons to the quinonate form, consistent with the formation of 4 upon treatment of 1a with aryl azide. Previous studies on nitrene transfer from organic azides to transition metals indicate that this process does not occur in a single step, but rather by initial coordination of the azide followed by nitrene transfer;34–36 however in this case, single step or multi-step nitrene transfer processes are kinetically indistinguishable. With step b as the rate determining step, the observed rate constant, kobs, is a composite of the equilibrium constant for step a (Ka) and the rate constant for step b (kb).
Steps c and d of Scheme 2 are migratory insertion and reductive elimination steps that reduce the ligand with concomitant formation of the carbodiimide CN bond. These steps are the key to turnover of the nitrene transfer reaction and they must occur faster than dimerization of the terminal imido complex to form 4. An alternative pathway to form carbodiimide would be through an intermolecular attack by free isonitrile on the zirconium imide group. Two factors suggest that such an intermolecular path is not operative. First, terminal imido ligands coordinated to early transition metals are typically nucleophilic37 and therefore should not react rapidly with free isonitrile (a nucleophile). Secondly, neither the bridging imido of complex 4, nor the terminal imido of the related tantalum complex, [NNNq]TaCl2(NPh), react with free isonitrile to form the carbodiimide product, even at elevated temperatures. The lack of reactivity of these coordinatively saturated imido complexes suggests that the isonitrile must be coordinated to the metal center prior to coupling with the imido ligand. This interpretation of the data implies umpolung reactivity in the nitrene transfer reaction. The imide, which is the oxidant, acts as the initial nucleophile, while the isonitrile, which is the reductant, acts as the initial electrophile. Nucleophilic attack of the imido ligand on coordinated isonitrile to form an η2-carbodiimide intermediate, step c, is consistent with previous reactivity studies between zirconium–imido complexes and isonitriles.32 In the present case, this idea is more intriguing since the putative η2-carbodiimide intermediate then releases the carbodiimide by reductive elimination (step d), sending two electrons back to the redox-active ligand.
Conclusions
The nitrene transfer reaction catalyzed by complex 1b illustrates the atypical reactivity engendered by the use of redox-active ligands and early transition metals. First, that 1b can act as a catalyst for nitrene transfer shows that the two-electron reactivity engendered by the [NNNcat]3− ligand is reversible, allowing the zirconium(IV) center both to accept a nitrene to form an imide species and subsequently to deliver the nitrene to an oxidizable substrate. The importance of reversibility in the nitrene transfer is highlighted by comparison to the only other examples of nitrene transfer reactivity from d0, group 4 metal imido complexes. In these studies, stoichiometric nitrene transfer from a titanium imide to either CO38 or CNR39 was observed, but the product metal fragment could only be stabilized in the presence of strong π-acceptor ligands, precluding catalytic turnover. In the case of 1b, the redox-active ligand can readily donate and accept two electrons, allowing catalytic turnover in a zirconium complex where the zirconium(II) oxidation state would be even more difficult to access than titanium(II).Another intriguing aspect of this nitrene transfer reaction is the strongly nucleophilic character of zirconium–imide in complex 1b. Nitrene transfer reactivity is well established for mid- and late-transition metal imido complexes of iron,40,41cobalt,42 and nickel;43 however, in late-transition-metal systems, the imide often acts as an electrophile in the initial attack on the substrate.11,44,45 In early-transition-metal complexes the imide is strictly nucleophilic, so for 1b, nitrene transfer can only occur to the isocyanide after it is activated by coordination to the electrophilic metal center. This contrasting reactivity to mid- and late-transition metal imido complexes may offer a means for different substrate selectivities in nitrene transfer catalysis.
The unusual aspects of this group-transfer reactivity provide motivation for further exploration into the coordination chemistry of early transition metals with redox-active ligands and hint at the potential for developing new kinds of catalytic coupling reactions.
Acknowledgements
This work was supported by the NSF-CAREER program (CHE-0645685) and the ACS Division of Organic Chemistry SURF program (to A. I. N.). A. F. H. is an Alfred P. Sloan Research Fellow and a Camille Dreyfus Teacher–Scholar.Notes and references
- W. A. Nugent and B. L. Haymore, Coord. Chem. Rev., 1980, 31, 123–175 CrossRef CAS
.
- D. E. Wigley, Prog. Inorg. Chem., 1994, 42, 239–482 CAS
- J. S. Johnson and R. G. Bergman, J. Am. Chem. Soc., 2001, 123, 2923–2924 CrossRef CAS
- A. L. Odom, Dalton Trans., 2005, 225–233 RSC
- C. Müller, W. Saak and S. Doye, Eur. J. Org. Chem., 2008, 2731–2739 CrossRef
- D. C. Leitch, P. R. Payne, C. R. Dunbar and L. L. Schafer, J. Am. Chem. Soc., 2009, 131, 18246–18247 CrossRef CAS
- H. M. Hoyt and R. G. Bergman, Angew. Chem., Int. Ed., 2007, 46, 5580–5582 CrossRef CAS
- C. P. Schaller, C. C. Cummins and P. T. Wolczanski, J. Am. Chem. Soc., 1996, 118, 591–611 CrossRef CAS
- J. L. Bennett and P. T. Wolczanski, J. Am. Chem. Soc., 1997, 119, 10696–10719 CrossRef CAS
- L. G. M. Slaughter, P. T. Wolczanski, T. R. Klinckman and T. R. Cundari, J. Am. Chem. Soc., 2000, 122, 7953–7975 CrossRef CAS
- P. Müller and C. Fruit, Chem. Rev., 2003, 103, 2905–2920 CrossRef CAS
- L. D. Amisial, X. Dai, R. A. Kinney, A. Krishnaswamy and T. H. Warren, Inorg. Chem., 2004, 43, 6537–6539 CrossRef CAS
- J. A. Halfen, Curr. Org. Chem., 2005, 9, 657–669 CrossRef CAS
- E. R. King and T. A. Betley, Inorg. Chem., 2009, 48, 2361–2363 CrossRef CAS
- Y. M. Badiei, A. Dinescu, X. Dai, R. M. Palomino, F. W. Heinemann, T. R. Cundari and T. H. Warren, Angew. Chem., Int. Ed., 2008, 47, 9961–9964 CrossRef CAS
- H. Y. Thu, W. Y. Yu and C. M. Che, J. Am. Chem. Soc., 2006, 128, 9048–9049 CrossRef CAS
- M. P. Jensen, M. P. Mehn and L. Que, Angew. Chem., Int. Ed., 2003, 42, 4357–4360 CrossRef CAS
- F. Ragaini, A. Penoni, E. Gallo, S. Tollari, C. Li Gotti, M. Lapadula, E. Mangioni and S. Cenini, Chem.–Eur. J., 2003, 9, 249–259 CrossRef
- M. Shen, B. E. Leslie and T. G. Driver, Angew. Chem., Int. Ed., 2008, 47, 5056–5059 CrossRef CAS
- E. Kogut, H. L. Wiencko, L. Zhang, D. E. Cordeau and T. H. Warren, J. Am. Chem. Soc., 2005, 127, 11248–11249 CrossRef CAS
- Y. M. Badiei, A. Krishnaswamy, M. M. Melzer and T. H. Warren, J. Am. Chem. Soc., 2006, 128, 15056–15057 CrossRef CAS
- J. S. Silvia and C. C. Cummins, J. Am. Chem. Soc., 2009, 131, 446–447 CrossRef CAS
- B. L. Tran, M. Pink, X. Gao, H. Park and D. J. Mindiola, J. Am. Chem. Soc., 2010, 132, 1458–1459 CrossRef CAS
- K. J. Blackmore, J. W. Ziller and A. F. Heyduk, Inorg. Chem., 2005, 44, 5559–5561 CrossRef CAS
- N. A. Ketterer, H. Fan, K. J. Blackmore, X. Yang, J. W. Ziller, M. H. Baik and A. F. Heyduk, J. Am. Chem. Soc., 2008, 130, 4364–4374 CrossRef CAS
- K. J. Blackmore, M. B. Sly, M. R. Haneline, J. W. Ziller and A. F. Heyduk, Inorg. Chem., 2008, 47, 10522–10532 CrossRef CAS
- K. J. Blackmore, N. Lal, J. W. Ziller and A. F. Heyduk, Eur. J. Inorg. Chem., 2009, 735–743 CrossRef CAS
- M. R. Haneline and A. F. Heyduk, J. Am. Chem. Soc., 2006, 128, 8410–8411 CrossRef CAS
- R. A. Zarkesh, J. W. Ziller and A. F. Heyduk, Angew. Chem., Int. Ed., 2008, 47, 4715–4718 CrossRef CAS
- A. I. Nguyen, K. J. Blackmore, S. M. Carter, R. A. Zarkesh and A. F. Heyduk, J. Am. Chem. Soc., 2009, 131, 3307–3316 CrossRef CAS
- R. R. Schrock, S. W. Seidel, Y. Schrodi and W. M. Davis, Organometallics, 1999, 18, 428–437 CrossRef CAS
- P. J. Walsh, F. J. Hollander and R. G. Bergman, Organometallics, 1993, 12, 3705–3723 CrossRef CAS
- D. J. Arney, M. A. Bruck, S. R. Huber and D. E. Wigley, Inorg. Chem., 1992, 31, 3749–3755 CrossRef CAS
- G. Proulx and R. G. Bergman, J. Am. Chem. Soc., 1995, 117, 6382–6383 CrossRef CAS
- M. G. Fickes, W. M. Davis and C. C. Cummins, J. Am. Chem. Soc., 1995, 117, 6384–6385 CrossRef CAS
- G. Proulx and R. G. Bergman, Organometallics, 1996, 15, 684–692 CrossRef CAS
- N. Hazari and P. Mountford, Acc. Chem. Res., 2005, 38, 839–849 CrossRef CAS
- T. E. Hanna, I. Keresztes, E. Lobkovsky, W. H. Bernskoetter and P. J. Chirik, Organometallics, 2004, 23, 3448–3458 CrossRef CAS
- S. C. Dunn, N. Hazari, A. R. Cowley, J. C. Green and P. Mountford, Organometallics, 2006, 25, 1755–1770 CrossRef CAS
- S. D. Brown, T. A. Betley and J. C. Peters, J. Am. Chem. Soc., 2003, 125, 322–323 CrossRef CAS
- R. E. Cowley, N. A. Eckert, J. Elhaïk and P. L. Holland, Chem. Commun., 2009, 1760–1762 RSC
- D. M. Jenkins, T. A. Betley and J. C. Peters, J. Am. Chem. Soc., 2002, 124, 11238–11239 CrossRef CAS
- D. J. Mindiola and G. L. Hillhouse, Chem. Commun., 2002, 1840–1841 RSC
- J. Du Bois, C. S. Tomooka, J. Hong and E. M. Carreira, Acc. Chem. Res., 1997, 30, 364–372 CrossRef CAS
- S. K. Leung, W. M. Tsui, J. S. Huang, C. M. Che, J. L. Liang and N. Zhu, J. Am. Chem. Soc., 2005, 127, 16629–16640 CrossRef CAS
Footnotes |
| † Electronic supplementary information (ESI) available: Detailed experimental procedures and complete characterization data for new complexes. CCDC reference number 780624. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c0sc00414f |
| ‡ Crystal data for 4: C64H88Cl2N8O5Zr2, M = 1302.76, orthorhombic, space groupP212121, a = 14.7192(7), b = 20.6103(9), c = 21.1775(9) Å, U = 6424.6(5) Å3, Z = 4, T = 88 K, μ = 0.461 mm−1, 57996 reflections collected, 15603 unique (Rint = 0.0419), Final R1 = 0.0376, GOF = 1.034, wR2 [I > 2σI] = 0.0829. |
| This journal is © The Royal Society of Chemistry 2011 |