Vinylogous reactivity of silver(I) vinylcarbenoids†
Jørn H.
Hansen
and
Huw M. L.
Davies
*
Department of Chemistry, Emory University, 1515 Dickey Drive, Atlanta, GA 30322, USA. E-mail: hmdavie@emory.edu; Fax: +1 404-727-7766; Tel: +1 404-727-6839
First published on 22nd November 2010
Abstract
Silver(I)-catalyzed reactions of vinyldiazoacetates have been shown to involve the intermediacy of silver-carbenoid species. The silver vinylcarbenoid displays a strong preference for electrophilic reactivity at the vinyl terminus in O–H insertion reactions.
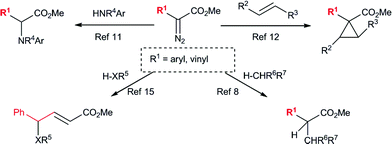
The application of silver salts as catalysts for decomposition of diazo compounds is traditionally associated with the venerable Wolff rearrangement.1 This transformation has found extensive use in synthesis.1–3 In contrast to copper and rhodium-catalysts, silver carbenoids are highly reactive and will often undergo rearrangement chemistry rather than reactions with external reagents. In recent years, silver-catalyzed reactions of diazo compounds have moved beyond this classical chemistry (Scheme 1). Diaz and Lovely,4–7 as well as Perez,8–10 have described the use of silver tris(pyrazolyl)borate complexes to catalyze carbenoid reactions with a variety of diazo compounds.4–10 A major advance in this area has been to combine silver catalysts with donor/acceptor carbenoids derived from aryl- and vinyldiazoacetates.7,11–13 These carbenoids are more stabilized than conventional carbenoids lacking a donor group14 and, consequently, are capable of reacting with external reagents under silver-catalyzed conditions. We have recently reported that aryl- and styryldiazoacetates can readily be decomposed by a catalytic amount of AgSbF6 to effect cyclopropanation reactions of sterically hindered alkenes that were unreactive using rhodium catalysis.12
 | ||
| Scheme 1 Ag(I)-catalyzed intermolecular reactions of aryl– and vinyldiazoacetates. | ||
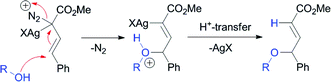
Silver-catalyzed O–H15 and N–H11insertion reactions have been reported with aryldiazoacetates; however, these reactions were considered to go via Lewis acid-catalyzed processes rather than carbenoid intermediates (Scheme 1).11,15 Hu and co-workers reported that 4-substitution could occur when methyl styryldiazoacetate was decomposed by silver salts and other Lewis acids in the presence of benzyl alcohol.15 This process was proposed to occur through a Lewis acid-catalyzed pathway (Scheme 2).15 In view of our previous studies, suggesting that silver-catalyzed reactions of aryl- and vinyldiazoacetates involve carbenoid intermediates,12 as well as our long-standing interest in vinylogous reactivity of vinylcarbenoids,16–18 we decided to conduct a more detailed investigation of this chemistry. Herein, we describe a comparative study of the silver(I)- and rhodium(II)-catalyzed reactions of vinyldiazoacetates, and demonstrate that high preference for vinylogous versuscarbenoid reactivity can be achieved under silver-catalyzed conditions. A computational analysis suggests that these reactions do indeed proceed through silver vinylcarbenoid intermediates.
 | ||
| Scheme 2 Proposed Lewis acid mechanism.15 | ||
We first re-investigated a system reported by Hu,15 the reaction between methyl styryldiazoacetate (1) and benzyl alcohol, catalyzed by AgOTf. The reaction can afford products 2 and 3, corresponding to vinylogous and carbenoid insertion, respectively (Scheme 3, Table 1). Although it was reported that exclusive formation of 2 occurs in refluxing dichloromethane with 10 mol% catalyst loading,15 we were unable to reproduce this result. Under these conditions, a product ratio of 85![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :15 was observed. The catalyst loading could be reduced to 5 mol % without affecting the yield or selectivity. At ambient temperature, the regioselectivity improved, but the best selectivity was obtained at 0 °C, which gave a 94:6 ratio in favor of 2 in 74% isolated yield.
:15 was observed. The catalyst loading could be reduced to 5 mol % without affecting the yield or selectivity. At ambient temperature, the regioselectivity improved, but the best selectivity was obtained at 0 °C, which gave a 94:6 ratio in favor of 2 in 74% isolated yield.
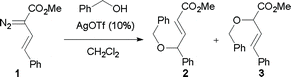 | ||
| Scheme 3 O–H insertion with benzyl alcohol. | ||
A standard test reaction that has been employed to evaluate vinylogous reactivity in vinylcarbenoids is the decomposition of 1 in neat methanol at ambient temperature (Scheme 4, Table 2).19 The reaction can give four products, two of which are derived from vinylogous reactivity (E and Z-4) and two from carbenoid reactivity (5 and 6). Isomers 5 and 6 have been shown to be favored with common dirhodium catalysts.19 However, with electron-deficient diruthenium(I) carbonyl carboxylates as catalysts, Z-4 was shown to be the favored product (75% selectivity).16 The same reaction with AgOTf displays strong selectivity for E-4 with 89% product selectivity, similar to the reactions with benzyl alcohol. The transformation was clean and proceeded in 95% combined yield of O–H insertion products.
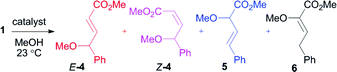 | ||
| Scheme 4 O–H insertion reaction in neat methanol. | ||
In order to further study the vinylogous insertion chemistry, we subjected several substrates to the reaction conditions using AgOTf and Rh2(OOct)4 as catalysts with diazo compounds 1 and 7 (Scheme 5, Table 3). A range of products 2, 3, 8a–h and 9a–g could be generated readily. Benzylic alcohols were good substrates for this chemistry, affording good regioselectivity for all cases in 64–74% yields. When employing benzyl amine, no reaction was observed with either the rhodium or silver-catalysts. This could be due to formation of stable metal-amine adducts. In the presence of aniline, however, a reaction was observed, but the regioselectivity was much attenuated in the case of the silver-catalyzed process, with only a 56:44 ratio in favor of the 4-substituted product 8b. In comparison, Hu reported a 65:35 ratio of products in 66% combined yield in refluxing dichloromethane for this reaction.15 The combined yields were good for both silver and rhodium (84%) catalyzed reactions. Good regioselectivity could also be observed when a carboxylic acid was employed as the substrate, and both rhodium and silver-catalysts gave 66% yields of 4- and 2-substituted products 8c and 9c, respectively. An allylic alcohol gave 67% of 8d with AgOTf, and 81% of carbenoid insertion 9d with Rh2(OOct)4. The homopropargylic alcohol gave an 81:19 ratio in favor of 4-substituted product 8e, when using the silver-catalyst. By using methyl (4-methoxy)styryldiazoacetate 7, a somewhat cleaner reaction was achieved with benzyl alcohol catalyzed by AgOTf, affording 77% of the 4-substituted product 8g in excellent regioselectivity. The electron-donating methoxy group may help to stabilize the electron-deficient silver carbenoid intermediate. The improved regioselectivity prompted a re-evaluation of aniline as the substrate, which gave a slightly improved regioselectivity (65:35 ratio).
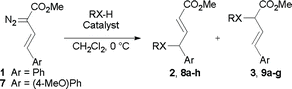 | ||
| Scheme 5 X–H insertions with arylvinyldiazoacetates. | ||
| Entry | Substrate | Prod. | Ar = | Catalyst | Ratio 8:9b | Yield (%)c |
|---|---|---|---|---|---|---|
| a A mixture of catalyst (0.01 equiv. for Rh and 0.05 equiv. for Ag), CH2Cl2 and substrate (2.1 equiv.), kept under a dry argon atmosphere, was cooled to 0 °C in an ice bath. The diazo compound (1.0 mmol, 1 equiv.), dissolved in CH2Cl2, was added drop-wise over 2 h after which the reaction mixture was allowed to slowly reach ambient temperature over 2-12 h. b From 1H NMR of crude reaction mixture. c Yield of major product after flash column chromatography. d Combined yield of both isomers. | ||||||
| 1 | PhCH2OH | 2 | -Ph | AgOTf | 94:6 |
74 |
| 2 | 3 | -Ph | Rh2(OOct)4 | <5:95 | 64 | |
| 3 | (4-NO2)C6H4CH2OH | 8a | -Ph | AgOTf | 94:6 |
64 |
| 4 | 9a | -Ph | Rh2(OOct)4 | <5:95 | 36 | |
| 5 | PhNH2 | 8b | -Ph | AgOTf | 56:44 |
84d |
| 6 | 9b | -Ph | Rh2(OOct)4 | <5:95 | 84 | |
| 7 | PhCH2COOH | 8c | -Ph | AgOTf | >95:5 |
66 |
| 8 | 9c | -Ph | Rh2(OOct)4 | <5:95 | 66 | |
| 9 |
|
8d | -Ph | AgOTf | 95:5 |
67 |
| 10 | 9d | -Ph | Rh2(OOct)4 | <5:95 | 81 | |
| 11 |
|
8e | -Ph | AgOTf | 81:19 |
41 |
| 12 | 9e | -Ph | Rh2(OOct)4 | <5:95 | 74 | |
| 13 |

|
8f | -Ph | AgOTf | 95:5 |
52 |
| 14 | 9f | -Ph | Rh2(OOct)4 | <5:95 | 64 | |
| 15 | PhCH2OH | 8g | -(4-MeO)Ph | AgOTf | >95:5 |
77 |
| 16 | 9g | -(4-MeO)Ph | Rh2(OOct)4 | <5:95 | 64 | |
| 17 | PhNH2 | 8h | -(4-MeO)Ph | AgOTf | 65:35 |
58d |
The vinylogous addition chemistry was also observed for other vinyldiazo compounds, such as the unsubstituted vinyldiazoacetate 10 (Scheme 6).19 A 49% yield of 13 could be isolated, but the reaction occurred with decreased selectivity compared to the O–H insertions of 1. The silver-catalyzed reaction was more selective than the Rh(II)-catalyzed process, as all three products 11–1319 were observed by 1H NMR of the crude reaction mixture in the latter case.
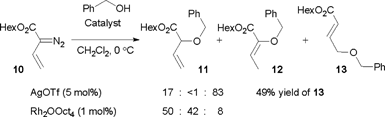 | ||
| Scheme 6 Insertion chemistry of diazo compound 10. | ||
The synthetic value of vinylogous reactivity would be greatly enhanced if regioselective C–C bond formation could occur. A previously employed test reaction for this is the reaction between unsubstituted vinyldiazoacetate 10 and N-Boc pyrrole (Scheme 7, Table 4).20 This could potentially produce a [4 + 3]-cycloadduct (carbenoid reactivity) or the 2-alkylation product 14 (vinylogous reactivity).20 With AgOTf as catalysts, 49% yield of 14 was isolated as a mixture of E and Z-isomers in a 82:18 ratio. In comparison, the reactions with rhodium(II)-catalysts prefer the Z-isomer, and only 33–42% yield was obtained with Rh2(OOct)4‡ and Rh2(TFA)4.20
 | ||
| Scheme 7 Vinylogous alkylation of N-Boc pyrrole. | ||
Considering the success of styryldiazoacetate 1 in the O–H insertion chemistry, vinylogous C–C bond formation was also attempted by silver-catalyzed decomposition in neat furan (Scheme 8). The reaction gave a good overall yield (80%), but relatively low selectivity between products 15–17 was observed. Only the minor product 15 is derived from vinylogous reactivity. The oxabicyclic product 16 was formed as the endo isomer, following the typical stereoselectivity of vinylcarbenoid reactions with furans.21 The trienal 17 is derived from a furan ring-opening reaction, consistent with previous findings in rhodium(II)-chemistry.21
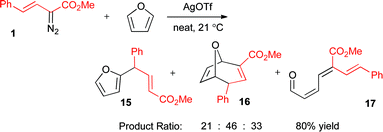 | ||
| Scheme 8 Reaction between 1 and furan. | ||
In order to investigate the likely mechanism of the silver-catalyzed transformations, we carried out reactions in MeOD (Scheme 9). Almost full incorporation of the deuterium label at the 2-position of the resulting butenoates was observed, consistent with selective proton transfer in the zwitterionic adducts.19 In the rhodium-catalyzed reaction, incorporation of the label in the 2-position is consistent with the currently accepted mechanism for rhodium carbenoid O–H insertions.22
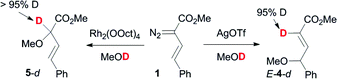 | ||
| Scheme 9 Deuterium labelling studies. | ||
A central issue in the silver(I)-catalyzed reactions of vinyldiazoacetates is whether the mechanism involves metallocarbenoids or proceeds through a Lewis acid-induced pathway.4,11,15,23 Although recent experimental evidence suggests that carbenoids are intermediates,12 there are, to our knowledge, no reports that demonstrate direct evidence for this.24 Therefore, it was decided to conduct a density functional study to explore whether a reaction pathway is viable through a silver vinylcarbenoid species. An evaluation of the Lewis acid catalysis mechanism by Hu et.al.15 on vinylogous X–H insertion was also conducted.
The reaction between styryldiazoacetate 1 and methanol was used as a model for the theoretical calculations. The main discussion is based on single-point energy calculations at the B3LYP/6-311+G(2d,2p)[Ag-RSC + 4f]//B3LYP/6-31G*[Ag-RSC + 4f]§ level of theory (see ESI†), with zero-point energy corrections and Gibbs free energies from B3LYP/6-31G*[Ag-RSC + 4f] calculations.
The potential energy surface for reaction between 1 and methanol, catalyzed by AgOTf, is shown in Fig. 1 along with structures of the stationary points (see ESI† for detailed geometries). Several adducts between AgOTf and 1 were explored, and it was found that the O-coordinated complex LA-I is the most stable adduct. This is consistent with the known Lewis-acidity profile of silver-salts.23 Complex LA-I is stabilized relative to free reactants by −20.5 kcal mol−1. The C-coordination complex LA-II was less stable (−12.7 kcal mol−1). However, a transition structure was found for loss of nitrogen from the latter, TS-II, which was only 5.65 kcal mol−1 less stable than LA-II. From LA-I, this corresponds to an overall potential energy barrier of +13.4 kcal mol−1. TS-II led to the silver(I)-vinylcarbenoid complex VC through an overall exoergic process (−35.6 kcal mol−1). For comparison, a transition structure was also found for loss of nitrogen gas from LA-I, but was disfavored over the vinylcarbenoid formation, as it displayed a potential energy barrier of +23.7 kcal mol−1. These results strongly suggest that formation of a silver vinylcarbenoid complex is a facile and highly exoergic process in this reaction system.
![Potential energy surface for silver-catalyzed carbenoid formation and trapping by methanol. Potential energies are reported relative to free reactants at the B3LYP/6-311+G(2d,2p)[Ag-RSC + 4f]//B3LYP/6-31G*[Ag-RSC + 4f] level of theory with zero-point corrections. Gibbs free energies are based on B3LYP/6-31G*[Ag-RSC + 4f] calculations.](/image/article/2011/SC/c0sc00422g/c0sc00422g-f1.gif) | ||
| Fig. 1 Potential energy surface for silver-catalyzed carbenoid formation and trapping by methanol. Potential energies are reported relative to free reactants at the B3LYP/6-311+G(2d,2p)[Ag-RSC + 4f]//B3LYP/6-31G*[Ag-RSC + 4f] level of theory with zero-point corrections. Gibbs free energies are based on B3LYP/6-31G*[Ag-RSC + 4f] calculations. | ||
The s-cis conformation of vinylcarbenoid complex VC was strongly favored over the s-trans orientation by ΔG = −5.39 kcal mol−1 (Scheme 10). In dirhodium vinylcarbenoid calculations, the energetic difference between the two is often small and slightly in favor of the s-trans isomer.25 In this regard, it appears that the triflate counterion is playing an important role by forming attractive interactions with the vinylic hydrogen adjacent to the phenyl group, which would stabilize the s-cis conformation. This explains why the vinylogous insertion products in the silver-catalyzed reactions are mainly formed with an E-double bond configuration.
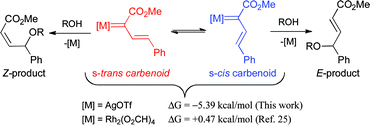 | ||
| Scheme 10 Influence of vinylcarbenoid conformations. | ||
Transition structures were next sought for the addition of methanol to both the vinylogous and carbenoid sites of VC. The two transition states TS-IIIa and TS-IIIb were located. A noteworthy characteristic of these structures was the preference for hydrogen bonding interactions between the O–H group and the triflate counterion. The two transition structures were next optimized at the B3LYP/6-311+G(d,p)[Ag-RSC + 4f] level of theory at 0 °C, with added solvent effect from CH2Cl2 (ε = 8.93) using the IEFPCM model, as implemented in Gaussian 09.26 The free energy difference between TS-IIIa and TS-IIIb was calculated to be ΔΔG‡ = 2.55 kcal mol−1, in reasonable agreement with data from Table 3 (regioisomer ratio from the reaction with benzyl alcohol indicates ΔΔG‡ ∼1.9 kcal mol−1), considering potential errors involved in DFT calculations.27 These studies show that vinylogous addition is the kinetically favored process.
The zwitterions ZW-IIa and ZW-IIb, resulting from addition to VC, would undergo rapid protonolysis to give the corresponding O–H insertion products. The proton transfer mechanism was not studied here. Transition structures that would correspond to the mechanism suggested by Hu15 could not be located. The Kohn–Sham LUMO of complex LA-I did not display any amplitude at the vinylogous position either. This is best illustrated by the LUMO frontier density plot shown in Fig. 2a, in which the blue area indicates the likely sites of electrophilic reactivity in the molecule. For LA-I this is located totally on the diazonium moiety. In comparison, Fig. 2b illustrates the same density surface for carbenoid complex VC. In this complex, electrophilic reactivity is displayed at both the carbenoid and vinylogous positions, consistent with previous experimental studies and the work presented herein.12,15
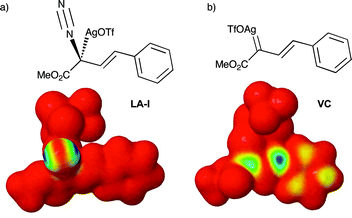 | ||
| Fig. 2 LUMO frontier density plots of: (a) LA-I and, (b) VC. | ||
Herein, we have demonstrated the ability of a silver(I) salt to enhance vinylogous addition chemistry in reactions with vinyldiazoacetates. The process is complementary to analogous reactions catalyzed by Ru(I)- or Rh(II)-complexes16,19 and the control of regioselectivity is effective for a variety of alcohols and even carboxylic acids in O–H insertions. C–C bond forming reactions occur with moderate yield and E/Z-selectivity. Density functional calculations have demonstrated that silver(I)-vinylcarbenoids are readily formed, and that they are likely intermediates in the vinylogous addition chemistry.
Financial support for this work by the National Science Foundation is gratefully acknowledged (CHE 0750273). We also wish to acknowledge Dr Jochen Autschbach for consultations and generously providing computational resources. The University at Buffalo's Center for Computational Research is acknowledged for technical support. Mr. Clay Owens is acknowledged for contributions to the optimization of the alkylation chemistry.
Notes and references
- W. Kirmse, Eur. J. Org. Chem., 2002, 2193–2256 CrossRef CAS.
- T. Honda, N. Haze, H. Ishige, K. Masuda, K. Naito and Y. Suzuki, J. Chem. Soc., Perkin Trans. 1, 1993, 539–540 RSC.
- P. Magnus and N. Westwood, Tetrahedron Lett., 1999, 40, 4659–4662 CrossRef CAS.
- H. V. R. Dias, R. G. Browning, S. A. Polach, H. V. K. Diyabalanage and C. J. Lovely, J. Am. Chem. Soc., 2003, 125, 9270–9271 CrossRef CAS.
- H. V. R. Dias, R. G. Browning, S. A. Richey and C. J. Lovely, Organometallics, 2004, 23, 1200–1202 CrossRef.
- C. J. Lovely, R. G. Browning, V. Badarinarayana and H. V. R. Dias, Tetrahedron Lett., 2005, 46, 2453–2455 CrossRef CAS.
- C. J. Lovely, J. A. Flores, X. F. Meng and H. V. R. Dias, Synlett, 2009, 129–132 CrossRef CAS.
- M. M. Díaz-Requejo and P. J. Pérez, J. Organomet. Chem., 2005, 690, 5441–5450 CrossRef CAS.
- J. Urbano, T. R. Belderrain, M. C. Nicasio, S. Trofimenko, M. M. Díaz-Requejo and P. J. Pérez, Organometallics, 2005, 24, 1528–1532 CrossRef CAS.
- J. Urbano, A. A. C. Braga, F. Maseras, E. Alvarez, M. M. Díaz- Requejo and P. J. Pérez, Organometallics, 2009, 28, 5968–5981 CrossRef CAS.
- S. Bachmann, D. Fielenbach and K. A. Jørgensen, Org. Biomol. Chem., 2004, 2, 3044–3049 RSC.
- J. L. Thompson and H. M. L. Davies, J. Am. Chem. Soc., 2007, 129, 6090–6091 CrossRef CAS.
- K. Burgess, H. J. Lim, A. M. Porte and G. A. Sulikowski, Angew. Chem., Int. Ed. Engl., 1996, 35, 220–222 CrossRef CAS.
- J. Hansen, J. Autschbach and H. M. L. Davies, J. Org. Chem., 2009, 74, 6555–6563 CrossRef CAS.
- Y. Yue, Y. Wang and W. Hu, Tetrahedron Lett., 2007, 48, 3975–3977 CrossRef CAS.
- Y. Sevryugina, B. Weaver, J. Hansen, J. L. Thompson, H. M. L. Davies and M. A. Petrukhina, Organometallics, 2008, 27, 1750–1757 CrossRef CAS.
- H. M. L. Davies, B. Hu, E. Saikali and P. R. Bruzinski, J. Org. Chem., 1994, 59, 4535–4541 CrossRef CAS.
- Y. Lian and H. M. L. Davies, Org. Lett., 2010, 12, 924–927 CrossRef CAS.
- H. M. L. Davies and Y. Yokota, Tetrahedron Lett., 2000, 41, 4851–4854 CrossRef CAS.
- H. M. L. Davies, E. Saikali and W. B. Young, J. Org. Chem., 1991, 56, 5696–5700 CrossRef CAS.
- S. J. Hedley, D. L. Ventura, P. M. Dominiak, C. L. Nygren and H. M. L. Davies, J. Org. Chem., 2006, 71, 5349–5356 CrossRef CAS.
- Y. Liang, H. L. Zhou and Z. X. Yu, J. Am. Chem. Soc., 2009, 131, 17783–17785 CrossRef CAS.
- M. Naodovic and H. Yamamoto, Chem. Rev., 2008, 108, 3132–3148 CrossRef CAS.
- H. V. R. Dias and C. J. Lovely, Chem. Rev., 2008, 108, 3223–3238 CrossRef CAS.
- D. T. Nowlan, T. M. Gregg, H. M. L. Davies and D. A. Singleton, J. Am. Chem. Soc., 2003, 125, 15902–15911 CrossRef CAS.
- Gaussian 09, Revision A.02. See ESI for the full reference.
- S. N. Pieniazek, F. R. Clemente and K. N. Houk, Angew. Chem., Int. Ed., 2008, 47, 7746–7749 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Procedures, characterization data, computational details, theoretical structures and selected spectral data are available. See DOI: 10.1039/c0sc00422g |
| ‡ In the reaction catalyzed by Rh2(OOct)4, a significant amount of product from reaction at the carbenoid site was observed (see ref. 20). |
| § The composite basis set consists of the 1997 Stuttgart RSC ECP for Ag, enhanced with two 4f-functions, and the split-valence basis set 6-31G* for the smaller atoms. For a similar basis set used in rhodium carbenoid chemistry, see ref. 14. A full account of the computational work is given in the ESI†. |
| This journal is © The Royal Society of Chemistry 2011 |