Metal-binding properties of Hpn from Helicobacter pylori and implications for the therapeutic activity of bismuth†
Seraphine V.
Wegner
,
Elif
Ertem
,
Murat
Sunbul
and
Chuan
He
*
Department of Chemistry and Institute for Biophysical Dynamics, The University of Chicago, 929 East 57th Street, Chicago, IL 60637, USA. E-mail: chuanhe@uchicago.edu
First published on 16th November 2010
Abstract
Nickel is of particular importance to Helicobacter pylori in part because it acts as a cofactor of urease, which is critical to the survival of H. pylori. In this study the nickel storage, histidine-rich protein Hpn from H. pylori was converted into a Ni2+ probe by inserting it between two fluorescence resonance energy transfer (FRET) partners, cyan fluorescent protein (CFP) and yellow fluorescent protein (YFP). The resulting construct, Hpn-FRET, exhibited a change in FRET upon the binding of Ni2+. Hpn-FRET has a moderate selectivity for Ni2+; it also responds to Zn2+ and Co2+ but not to other biometals. Competition experiments between Ni2+ and other metals plus the measured Kd values for Zn2+ and Ni2+ establish the selectivity order for Hpn-FRET as Zn2+ > Ni2+ > Co2+ ≫ other biometals. Bismuth is widely used as a therapeutic agent against H. pylori, and Hpn has been suggested as one of the possible targets. The dissociation constant of Bi3+ to Hpn-FRET was measured to be 6.19 × 10−5 M. Further experiments using Hpn-FRET in E. coli indicate that Hpn-FRET responds to Bi3+ but not to Ni2+ and Zn2+ inside E. coli. The result shows that unlike Ni2+ and Zn2+, which are tightly regulated in most bacteria, available Bi3+ can reach high micromolar levels inside E. coli.
Introduction
Helicobacter pylori is a gram-negative pathogenic bacterium which colonizes the digestive track and causes chronic gastritis, ulcers, and even some gastric cancers.1 This bacterium has the remarkable ability to survive in the hostile environment of the stomach and is found in about half the world's human population.2 Its ability to adapt to the acidic living conditions of the stomach lies in its high activity of urease, which catalyzes the hydrolysis of urea to produce carbon dioxide and ammonia that neutralize the internal pH.3Urease, accounting for about 10% of the total protein content in the bacterium, uses two nickel ions for catalysis in its holo form.4 Another important enzyme for H. pylori that also uses a nickel cofactor is hydrogenase which provides energy to this bacterium.5 Therefore, nickel is an essential metal for the survival of H. pylori, and its metabolism is globally regulated by the transcriptional regulatorHpNikR.6 An excess of nickel has been shown to be harmful, however, and the molecular mechanism of this toxicity is not understood in detail.One line of treatment used against H. pylori are bismuth-containing therapeutics such as bismuth subsalicylate, bismuth subcitrate, and ranitidine bismuth citrate.7 While the exact mechanism of action for bismuth remains unknown, all current hypotheses suggest binding of bismuth to the metal-binding sites in various metalloproteins.8In vitro studies have shown that Bi3+ can bind to the active sites of urease9 and alcohol dehydrogenase,10 resulting in the inhibition of the catalytic activity of these enzymes. Other proposed target proteins of Bi3+ are metallothioneins11 and the histidine-rich protein Hpn.12 While Bi3+ has a low toxicity in humans, it is known to interact with serum proteins such as transferrin,13 lactoferrin,14 and serum albumin.15
In H. pylori as well as many other organisms, transition metals such as nickel play a dual role: they are at once essential as cofactors to enzymes that catalyze important transformations, but are also toxic at elevated levels. Substantial work has been done to understand the elaborate homeostatic networks for these metals, the individual regulation, uptake, distribution, delivery, storage, efflux, and the role these metals play in various diseases; however, the picture is still far from complete.16,17 As part of the effort to elucidate the function of metals in living systems, fluorescent small-molecule and protein-based sensors have been developed to visualize metals.18 These two classes of sensors have complementary applications and advantages. While both small molecule and protein-based sensors for Ca2+,19,20Zn2+,21,22 and Cu+23,24 have been developed and successfully used in vivo, small molecule and protein-based probes for Ni2+ and other transition metals are still lacking.25
In this study Hpn, which is a histidine-rich protein, was converted into a FRET-based fluorescent sensor, Hpn-FRET, by inserting Hpn between the two FRET partners CFP (cyan fluorescent protein) and YFP (yellow fluorescent protein) (Fig. 1b). This approach to construct a protein based FRET sensor by introducing a metal binding domain which undergoes a conformational change between two FRET partners has been widely used19,21,26 and we have previously converted metallothioneins which are small cysteine-rich proteins into effective FRET probes that demonstrate high selectivity to copper(I) due to the assembly of a tetracopper(I) cluster.23 Hpn was chosen as a sensory domain because it has been reported to function as a nickel storage protein that provides nickel to the active site of urease in H. pylori.27,28 Similarly to metallothioneins Hpn bears a large number of coordinating amino acids in a short sequence and changes conformation upon metal binding as has been observed by CD.29,30 Overexpression of Hpn protects H. pylori from high concentrations of metal, especially nickel, and its expression is upregulated by HpNikR in the presence of excess Ni2+.29,30 Hpn has also been proposed as a target for bismuth because deletion of the hpngene decreases the survival of H. pylori with bismuth treatment.12 Hpn, is a histidine-rich small protein that contains 28 histidines out of a total of 60 residues (Fig. 1a) and has been shown to bind approximately 5 equiv. of Ni2+ (Kd = 7.1 × 10−6 M), 5 equiv. of Zn2+ (Kd = 19.28 × 10−6 M), 8.5 equiv. of Cu2+ (Kd = 2.16 × 10−6 M), and 3.81 equiv. of Bi3+ (Kd = 11.1 × 10−6 M), in a study in which the dissociation constants were determined by equilibrium dialysis.31
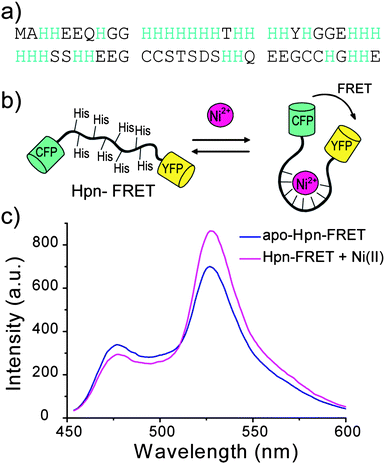 | ||
| Fig. 1 Design of Hpn-FRET. (a) Sequence of Hpn, which is a histidine-rich metallothionein. (b) Hpn sequence is inserted between the FRET partners CFP and YFP. The binding of metals to Hpn results in a conformational change which leads to a change of FRET signal. (c) Fluorescence response of Hpn-FRET to Ni2+. Apo Hpn-FRET is shown in blue, and Hpn-FRET with 6 equiv. Ni2+ is shown in pink. | ||
In the presented work, the metal binding properties of the resulting probe, Hpn-FRET, for different biometals and Bi3+ were characterized in vitro and the selectivity and affinity of Hpn-FRET were established. When Hpn-FRET was expressed in E. coli which is used as a model system, FRET change was only observed with the addition of Bi3+, but not Ni2+ and Zn2+. These measurements show that Hpn is a likely target for Bi3+in vivo. Unlike Ni2+ and Zn2+, which are tightly controlled in bacterial cells, the free concentration of Bi3+ can reach high micromolar levels.
Results
Design of Hpn-FRET and the response to Ni2+
Hpn is a histidine-rich protein and has been shown to bind to transition metals, resulting in a change in secondary structure (Fig. 1a). Taking advantage of this conformational change upon metal binding, we cloned Hpn between the two FRET partners CFP and YFP to yield Hpn-FRET (Fig. 1b). Hpn-FRET was overexpressed in E. coli, purified by Ni-NTA and gel filtration columns, and its purity was confirmed by a denaturing SDS-PAGE protein gel (Fig. S1, ESI†). Fluorescence spectra of Hpn-FRET (250 nM) were recorded in the absence and presence of 1.5 μM Ni2+ by exciting CFP (433 nm) and scanning from 450 to 600 nm. The addition of Ni2+ to Hpn-FRET resulted in a decrease in the fluorescence intensity of CFP (477 nm) and an increase in fluorescence intensity of YFP (527 nm) due to a change in the FRET efficiency upon metal binding (Fig. 1c). The conformational change of Hpn-FRET upon metal binding was also observed in the CD spectrum (Fig. S2, Table S1, ESI†).Selectivity of Hpn-FRET
The metal-binding properties of Hpn-FRET were investigated in detail by titrating Hpn-FRET with different biologically relevant metal ions. Titration of Hpn-FRET with Ni2+, the proposed metal that binds Hpn, showed 5 equiv. binding with a quite noticeable change in FRET (∼ 40%). Beside Ni2+, the addition of Zn2+ or Co2+ also resulted in a change in FRET. The FRET change with Zn2+ is larger than that observed with Ni2+ (∼55%), and the FRET change with Co2+ is smaller than that with Ni2+ (∼22%) (Fig. 2, Fig. S3, ESI†). Additionally when the CD spectra of Hpn-FRET in the presence of these metals were taken an increase in α-helix and a decrease in β-sheet content were observed where Zn2+ resulted in the biggest change and Co2+ in the least showing the same trend as the FRET response (Fig. S2, Table S1, ESI†). The titration also indicated that Hpn-FRET binds 5 equiv. of Zn2+, but only 4 equiv. of Co2+. A small change in FRET was observed upon addition of Cu2+ or Bi3+. Almost no FRET change was observed with Mg2+, Ca2+, Cr3+, Mn2+, Fe2+ or Fe3+ (Fig. 2).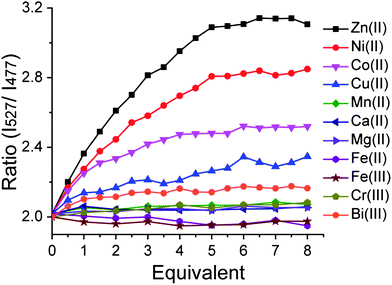 | ||
| Fig. 2 Hpn-FRET titration with different metal ions. Hpn-FRET (250 nM) was titrated with Zn2+, Ni2+, Co2+, Cu2+, Mn2+, Ca2+, Mg2+, Fe2+, Fe3+, Cr3+ and Bi3+ up to 8 equiv. | ||
The selectivity of Hpn-FRET for Ni2+ was further evaluated in competition experiments in which the response of Hpn-FRET to Ni2+ was measured in the presence of an equal amount of the competing metal ions Mg2+, Ca2+, Cr3+, Mn2+, Fe2+, Co2+, Cu2+, Zn2+, Fe3+ and Bi3+. In these experiments, first the response of Hpn-FRET was measured in the presence of 6 equiv. metal ions. Then, an equal amount of Ni2+ was added to the sample, and the response was measured again (Fig. 3). In all cases except for Zn2+, the full response to Ni2+ was observed, indicating that Ni2+ binds tighter than other metal ions and the presence of other metal ions does not disturb the Ni2+ binding. When the same experiments were done with Zn2+ or Co2+, again none of the other metal ions could interfere with the binding. Zn2+ was able to displace Ni2+ and Co2+, while Co2+ displaced neither Zn2+ nor Ni2+ (Fig. S4a,b, ESI†). Furthermore, the binding stoichiometry of 5 equiv. Ni2+, 5 equiv. Zn2+ and 4 equiv. Co2+ to Hpn-FRET was confirmed by submitting Hpn-FRET samples, incubated with 10 equiv. of the respective metal and separated from the unbound metal, for ICP-MS analysis (Table S2, ESI†). In agreement with the selectivity observed in the fluorescence measurements, when Hpn-FRET was incubated with two metals at the same time Hpn-FRET preferentially bound to Zn2+ over Ni2+ and Co2+, and to Ni2+ over Co2+ (Table S2, ESI†). From these competition experiments, a relative binding order of Zn2+ > Ni2+ > Co2+ ≫ other metals, can be obtained.
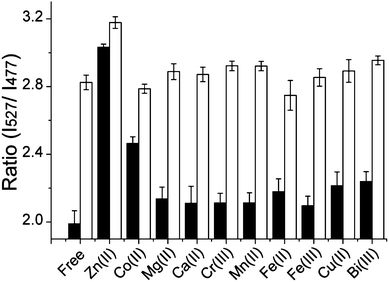 | ||
| Fig. 3 Competition of Ni2+ with other metal ions for binding to Hpn-FRET. Hpn-FRET (250 nM) with 6 equiv. of the respective metal ions is shown in black, and the addition of 6 equiv. Ni2+ to the sample is shown in white. All measurements were done in triplicates. | ||
K d measurements of Hpn-FRET for Zn2+ and Ni2+
To quantify the absolute binding affinity of Hpn-FRET to Ni2+ and Zn2+, binding constants were determined. Free Ni2+ concentrations were buffered from 1.37 × 10−9 to 6.32 × 10−6 M using various amounts of Ni2+ and citrate.32,33 The percentage of metal loading to Hpn-FRET was measured in these buffers and the binding curve was fitted using the Hill equation (Fig. 4a, Table S3, ESI†). A dissociation constant of Hpn-FRET to Ni2+ was computed to be 7.89 × 10−8 ± 8.9 × 10−9 M with no cooperativity observed (n = 1.21 ± 0.16). The dissociation constant for Zn2+ was measured similarly by buffering free Zn2+ from 3.94 × 10−11 to 6.64 × 10−7 M using Zn2+ and cyanide.32,33 From fitting the binding curve, a dissociation constant of 1.03 × 10−9 ± 9.3 × 10−11 M was obtained with no cooperativity observed (n = 1.42 ± 0.22) (Fig. 4b, Table S3, ESI†). These experiments indicate that Hpn-FRET has a high affinity for Ni2+ and an even higher (75-fold as compared to Ni2+) affinity for Zn2+.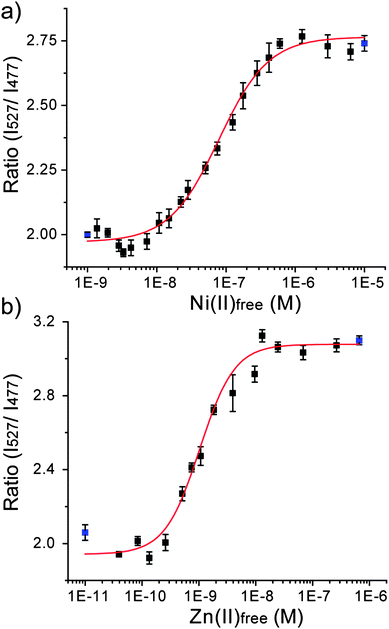 | ||
| Fig. 4 Binding curve of Hpn-FRET to (a) Ni2+ and (b) Zn2+. The fitting curve is shown in red and the blue points represent the minimum (apo-Hpn-FRET) and maximum (Hpn-FRET + 6 equiv. Ni2+ or + 6 equiv. Zn2+) signals, respectively. All measurements were done in triplicates. | ||
K d measurements of Hpn-FRET for Bi3+
We were puzzled by the low FRET change observed for Bi3+ shown in Fig. 2, which seems to be inconsistent with the proposal that Hpn in H. pylori is a target of Bi3+. Therefore, we set out to measure the binding constant for Bi3+ by adding increasing concentrations of Bi3+ in 10 μM increments to Hpn-FRET (250 nM). To our surprise, we observed a significant change of the FRET ratio from 2.0 to 4.0 at higher concentrations of Bi3+, and this increase in FRET was saturated at ∼90 μM Bi3+ (Fig. 5). The binding curve was fitted to the Hill equation and the dissociation constant was found to be 6.19 × 10−5 ± 1.0 × 10−6 M with cooperativity (n = 6.23 ± 0.71) (Table S3, ESI†). The Hill equation will only give an accurate number of binding sites when very strong cooperativity is present between the first and subsequent binding sites. It gives a lower limit to the number of binding sites in the case of moderate cooperativity.34 For the binding curves of Ni2+ and Zn2+, where no cooperativity is observed (n ∼ 1), the binding equivalence was determined separately and found to be 5 equiv. of the respective metal. In the case of Bi3+, n = 6.23 was obtained from the fitting of the Hill equation, and therefore, there are at least six metal binding sites with some cooperativity.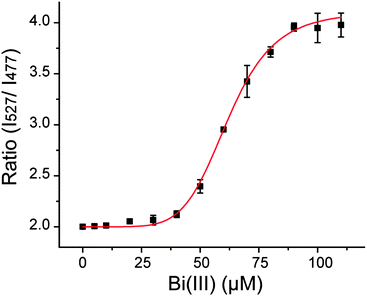 | ||
| Fig. 5 Binding curve of Hpn-FRET to Bi3+. The fitting curve is shown in red. All measurements were done in triplicate. | ||
Response of Hpn-FRET to Bi3+, Zn2+ and Ni2+ in E. coli
To evaluate the ability of Hpn-FRET to function as a probein vivo, Hpn-FRET was expressed in E. coli and FRET changes were measured when cells were incubated with Ni2+, Zn2+ or Bi3+.35Bismuth subsalicylate was used as a bismuth source because it is the active ingredient in Pepto Bismol, a common treatment for H. pylori.Pepto Bismol was used as well for comparison. E. colicells in MOPS minimal medium expressing Hpn-FRET were incubated with 1 to 400 μM metal complexes after protein expression was induced with 0.1 mM IPTG and cells grown overnight. To ensure that the metals did not inhibit growth, the OD600 was measured and no effect was observed. The fluorescence spectra of these cells were taken and the fluorescent energy transfer was quantified by taking the intensity ratio of the YFP emission (527 nm) over the CFP emission (477 nm). We did not observe a change of the FRET ratio when cells were incubated with Zn2+ or Ni2+, as compared to the control cells with no metal added (Fig. 6). On the other hand, addition of bismuth subsalicylate or Pepto Bismol resulted in a significant increase of the FRET ratio starting at 10–20 μM and saturating at ∼100 μM Bi3+ (Fig. 6, Fig. S5, ESI†). These results demonstrate that Hpn-FRET can efficiently bind to Bi3+ in the bacterial cellular environment, and that free Ni2+ and Zn2+ concentrations inside E. coli are not high enough for Hpn-FRET to respond to them.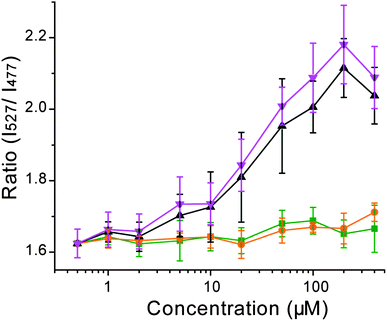 | ||
| Fig. 6 Response of Hpn-FRET inside E. coli to Ni2+ (orange), Zn2+ (green), bismuth subsalicylate (black) and Pepto Bismol (pink) (the total concentration of bismuth is shown in the x-axis). Cells were excited at 420 nm and the ratio of the intensities at 527 nm over 477 nm was plotted as response. Error bars represent the standard deviation from four independent cell cultures. | ||
Discussion
Hpn-FRET was designed by inserting Hpn between the FRET partners CFP and YFP, taking advantage of the previously reported change in Hpn conformation upon Ni2+ binding.29 The resulting Hpn-FRET serves as an excellent probe to study metal binding to Hpn while avoiding potential problems of protein aggregation and denaturation. Hpn-FRET shows a moderate selectivity for Ni2+; it also responds to Zn2+ and Co2+ but not other biometals, such as Mg2+, Ca2+, Cr3+, Mn2+, Fe2+, Fe3+ and Cu2+. Competition experiments give a relative binding order of Zn2+ > Ni2+ > Co2+ for Hpn. The measured binding constant of Hpn-FRET to Ni2+ is 7.89 × 10−8 M while the binding constant of Hpn-FRET to Zn2+ is 1.03 × 10−9 M, which is 75-fold tighter than that to Ni2+.The metal-binding properties of Hpn-FRET show that it binds 5 equiv. Ni2+ with a Kd = 7.89 × 10−8 M, 5 equiv. Zn2+ with a Kd = 1.03 × 10−9 M, 4 equiv. of Co2+ with an affinity lower than that of Ni2+ or Zn2+, and no appreciable binding to the other bioavailable metals Mg2+, Ca2+, Cr3+, Mn2+, Fe2+, Fe3+ and Cu2+. In addition, binding to the therapeutic metal Bi3+ is observed at a higher concentration with Kd = 6.19 × 10−5 M. Thus, the affinity order of Hpn-FRET to biometals is Zn2+ > Ni2+ > Co2+ > other metals tested and Bi3+. The metal-binding behavior of Hpn-FRET is different from previous reports that used the isolated Hpn peptide. While the binding stoichiometry of Hpn-FRET for Ni2+ and Zn2+ are in agreement with the Hpn peptide, Hpn-FRET binds much tighter to these two metal ions than previously reported using equilibrium dialysis and analyzing metal content by ICP-MS.29,31 Furthermore, binding of ∼8 equiv. of Cu2+ and ∼4 equiv. Bi3+ were not observed for Hpn-FRET. One potential explanation for this difference in the metal-binding behavior could be the difference in oligomerization of the Hpn peptide and Hpn-FRET. The Hpn peptide can form different large multimers depending on the buffer conditions.29 Hpn-FRET is less prone to aggregation as it elutes as one peak on a size-exclusion column in the presence and absence of Ni2+ (Fig. S6, ESI†). In a different study reporting the relative binding affinity of Hpn peptide to various metals using western blots in combination with radioisotopes, an affinity order of Ni2+ and Zn2+ ≫ Co2+ > Cu2+ > Mn2+ was reported,28 which is in agreement with what we observed with Hpn-FRET. Because the FRET technique is sensitive, and therefore less concentrated samples were used, as well as the presence of CFP and YFP at the two termini, multimerization of Hpn can be avoided.
Hpn has been suggested as a target for the therapeutic metal Bi3+ based on the higher susceptibility of the hpn knock-out strain of H. pylori to bismuth treatment.12 The much higher binding affinities of Hpn to Ni2+ and Zn2+ over Bi3+ pose the question of how Bi3+ can target Hpn in H. pylori. Therefore, the FRET response of Hpn-FRET inside a model bacterium, E. coli, towards Ni2+, Zn2+, Bi3+ and Pepto Bismol was evaluated. In these experiments, up to 400 μM metals were added to bacterial cells and no toxicity was observed. While the addition of Ni2+ or Zn2+ resulted in no increase in FRET compared to control cells, an increase of FRET was observed following incubation with either Pepto Bismol or bismuth subsalicylate, beginning with 10–20 μM Bi3+ and saturating at ∼100 μM Bi3+. This result shows that Hpn-FRET binds to Bi3+ efficiently inside E. coli, and that Hpn-FRET does not bind to Ni2+ or Zn2+ tightly enough to compete for these metals inside E. coli. While metal homeostasis in H. pylori and E. coli is clearly different it is intriguing to observe such a high accumulation of Bi3+ inside E. coli considering the therapeutic activity of Bi3+ against H. pylori. However, one should be cautious to extrapolate results obtained from experiments performed in E. coli to H. pylori. The nickel content in H. pylori could be significantly higher than that in E. coli, which could make Hpn predominantly occupied with nickel in H. pylori. To test the hypothesis experiments with Hpn-FRET in H. pylori would be very interesting for future studies.
It has been well established that most bacterial cells tightly control metal availability.16 In E. coli, nickel availability is limited to 10−12 M, which has been determined by the study of the Ni2+ -dependent transcriptional regulator NikR.36 The free zinc level in E. coli is likewise regulated to ∼10−15 M as was determined by the study of the zinc regulatory proteins Zur and ZntR.37 Since the Kd values of Hpn-FRET to Ni2+ and Zn2+ are 7.89 × 10−8 M and 1.03 × 10−9 M, respectively, they are unable to compete for these metals inside E. coli, which agrees with the absence of any FRET change in our experiments with the use of Hpn-FRET.
In H. pylori the Ni2+ level is controlled by HpNikR,38 which has been reported to bind Ni2+ at ∼1.2 × 10−8 M (tighter binding was also suggested depending on techniques used to determine the Kd value).39,40 This Kd value of HpNikR agrees very well with our measured Hpn-FRET Kd to Ni2+ of 7.89 × 10−8 M, supporting the hypothesis that Hpn acts as a storage protein to scavenge excess Ni2+ inside H. pylori. The previously reported Hpn peptideKd of 7.1 × 10−6 M to Ni2+ is too high to be consistent with Hpn acting as a be a nickel storage and scavenger protein considering the response threshold of HpNikR. On the other hand, Bi3+ is not regulated in bacterial cells and no specific detoxification mechanism for Bi3+ is known. Bismuth thus can accumulate to a high cellular concentration and occupy available metal-binding sites, interfering with biological functions. In fact, the suggested single dose for Pepto Bismol is high enough to result in ∼700 μM Bi3+ in the stomach, which is higher than the concentration needed to saturate Hpn-FRET inside E. coli. Our measurements in E. coli show that free, available Bi3+ can accumulate to high micromolar levels. If Bi3+ accumulates to the similarly high levels inside H. pylori it may target Hpn or related proteins, eventually leading to bactericidal effects. The exact consequence of bismuth binding to Hpn still remains to be evaluated, but it has been found that the hpn deficient H. pylori cannot adapt to decreases in pH of the growth medium and has lower urease activity when Ni2+ in the growth media was chelated. Thus, if binding of Bi3+ prevents Hpn function, the bacteria would be more susceptible to changing environmental conditions.
Conclusions
In closing, the histidine-rich protein Hpn from H. pylori, which functions as a Ni2+ storage protein and is a target for Bi3+ therapy, is converted into a FRET-based metal probe, Hpn-FRET. Taking advantage of the metal-binding-induced FRET change of this construct, we determined the metal-binding properties of this protein to be Zn2+ (Kd = 1.03 × 10−9 M) > Ni2+ (Kd = 7.89 × 10−8 M) > Co2+ > other biometals. In addition, the dissociation constant for the therapeutic metal Bi3+ to Hpn-FRET was measured to be 6.19 × 10−5 M. Experiments in E. coli showed that Ni2+ and Zn2+ can not bind to Hpn-FRET due to their tight regulation inside the cell; however, high micromolar levels of Bi3+ accumulate and result in the FRET change of Hpn-FRET inside E. coli. The FRET-based probe is an excellent tool to study sensitive binding of metal ions to metalloproteins, and can be used to probe metal availability in living cells.Acknowledgements
This work was supported by supported by a CAREER award from the National Science Foundation (NSF0544546 to C. H.) and Office of Basic Energy Sciences, U. S. Department of Energy under Contract No. DE-FG02-07ER15865 (to C. H.)Notes and references
- M. J. Blaser and J. C. Atherton, J. Clin. Invest., 2004, 113, 321 CAS.
- T. L. Cover and M. J. Blaser, Adv. Int. Med., 1996, 41, 85 Search PubMed.
- K. Stingl and H. De Reuse, Int. J. Med. Microbiol., 2005, 295, 307 CrossRef CAS.
- R. J. Maier, S. L. Benoit and S. Seshadri, Biometals, 2007, 20, 655 Search PubMed.
- S. L. Benoit and R. J. Maier, Ann. N. Y. Acad. Sci., 2008, 1125, 242 CrossRef CAS.
- (a) F. D. Ernst, E. J. Kuipers, A. Heijens, R. Sarwari, J. Stoof, C. W. Penn, J. G. Kusters and A. H. van Vliet, Infect. Immun., 2005, 73, 7252 CrossRef CAS; (b) N. S. Dosanjh and S. L. Michel, Curr. Opin. Chem. Biol., 2006, 10, 123 CrossRef CAS.
- (a) U. Dittes, E. Vogel and B. K. Keppler, Coord. Chem. Rev., 1997, 163, 345 CrossRef CAS; (b) P. J. Sadler, H. Li and H. Sun, Coord. Chem. Rev., 1999, 185–186, 689 CrossRef CAS; (c) N. Yang and H. Sun, Coord. Chem. Rev., 2007, 251, 2354 CrossRef CAS; (d) H. Sun, L. Zhang and K. Y. Szeto, Met. Ions Biol. Syst., 2004, 41, 333 CAS.
- R. Ge and H. Sun, Acc. Chem. Res., 2007, 40, 267 CrossRef CAS.
- L. Zhang, S. B. Mulrooney, A. F. Leung, Y. Zeng, B. B. Ko, R. P. Hausinger and H. Sun, BioMetals, 2006, 19, 503 Search PubMed.
- L. Jin, K. Y. Szeto, L. Zhang, W. Du and H. Sun, J. Inorg. Biochem., 2004, 98, 1331 CrossRef CAS.
- (a) H. Sun, H. Li, I. Harvey and P. J. Sadler, J. Biol. Chem., 1999, 274, 29094 CrossRef CAS; (b) J. Pannequin, S. Kovac, J. P. Tantiongco, R. S. Norton, A. Shulkes, K. J. Barnham and G. S. Baldwin, J. Biol. Chem., 2003, 279, 2453 CrossRef.
- H. L. Mobley, R. M. Garner, G. R. Chippendale, J. V. Gilbert, A. V. Kane and A. G. Plaut, Helicobacter, 1999, 4, 162 CrossRef CAS.
- H. Li, P. J. Sadler and H. Sun, J. Biol. Chem., 1996, 271, 9483 CrossRef CAS.
- L. Zhang, K. Y. Szeto, W. B. Wong, T. T. Loh, P. J. Sadler and H. Sun, Biochemistry, 2001, 40, 13281 CrossRef CAS.
- H. Sun, H. Li, A. B. Mason, R. C. Woodworth and P. J. Sadler, J. Biol. Chem., 2001, 276, 8829 CrossRef CAS.
- K. J. Waldron and N. J. Robinson, Nat. Rev. Microbiol., 2009, 7, 25 Search PubMed.
- K. J. Waldron, J. C. Rutherford, D. Ford and N. J. Robinson, Nature, 2009, 460, 823 CrossRef CAS.
- (a) D. W. Domaille, E. L. Que and C. J. Chang, Nat. Chem. Biol., 2008, 4, 168 CrossRef CAS; (b) E. L. Que, D. W. Domaille and C. J. Chang, Chem. Rev., 2008, 108, 1517 CrossRef CAS.
- A. Miyawaki, J. Llopis, R. Heim, J. M. McCaffery, J. A. Adams, M. Ikura and R. Y. Tsien, Nature, 1997, 388, 882 CrossRef CAS.
- M. Mank, A. F. Santos, S. Direnberger, T. D. Mrsic-Flogel, S. B. Hofer, V. Stein, T. Hendel, D. F. Reiff, C. Levelt, A. Borst, T. Bonhoeffer, M. Hubener and O. Griesbeck, Nat. Methods, 2008, 5, 805 CrossRef CAS.
- (a) J. L. Vinkenborg, T. J. Nicolson, E. A. Bellomo, M. S. Koay, G. A. Rutter and M. Merkx, Nat. Methods, 2009, 6, 737 CrossRef CAS; (b) W. Qiao, M. Mooney, A. J. Bird, D. R. Winge and D. J. Eide, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 8674 CrossRef CAS; (c) P. J. Dittmer, J. G. Miranda, J. A. Gorski and A. E. Palmer, J. Biol. Chem., 2009, 284, 16289 CrossRef CAS.
- (a) C. J. Chang, E. M. Nolan, J. Jaworski, S. C. Burdette, M. Sheng and S. J. Lippard, Chem. Biol., 2004, 11, 203 CrossRef CAS; (b) E. M. Nolan, J. W. Ryu, J. Jaworski, R. P. Feazell, M. Sheng and S. J. Lippard, J. Am. Chem. Soc., 2006, 128, 15517 CrossRef CAS.
- S. V. Wegner, H. Arslan, M. Sunbul, J. Yin and C. He, J. Am. Chem. Soc., 2010, 132, 2567 CrossRef CAS.
- (a) L. Zeng, E. W. Miller, A. Pralle, E. Y. Isacoff and C. J. Chang, J. Am. Chem. Soc., 2006, 128, 10 CrossRef CAS; (b) L. Yang, R. McRae, M. M. Henary, R. Patel, B. Lai, S. Vogt and C. J. Fahrni, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 11179 CrossRef CAS.
- (a) S. C. Dodani, Q. He and C. J. Chang, J. Am. Chem. Soc., 2009, 131, 18020 CrossRef CAS; (b) T. Mizuno, K. Murao, Y. Tanabe, M. Oda and T. Tanaka, J. Am. Chem. Soc., 2007, 129, 11378 CrossRef CAS.
- J. Zhang, Y. Ma, S. S. Taylor and R. Y. Tsien, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 14997 CrossRef CAS.
- S. Seshadri, S. L. Benoit and R. J. Maier, J. Bacteriol., 2007, 189, 4120 CrossRef CAS.
- J. V. Gilbert, J. Ramakrishna, F. W. Sunderman, Jr, A. Wright and A. G. Plaut, Infect. Immun., 1995, 63, 2682 CAS.
- R. Ge, R. M. Watt, X. Sun, J. A. Tanner, Q. Y. He, J. D. Huang and H. Sun, Biochem. J., 2006, 393, 285 CrossRef CAS.
- Y. B. Zeng, D. M. Zhang, H. Li and H. Sun, JBIC, J. Biol. Inorg. Chem., 2008, 13, 1121 CrossRef CAS.
- R. Ge, Y. Zhang, X. Sun, R. M. Watt, Q. Y. He, J. D. Huang, D. E. Wilcox and H. Sun, J. Am. Chem. Soc., 2006, 128, 11330 CrossRef CAS.
- L. Alderighi, P. Gans, A. Ienco, D. Peters, A. Sabatini and A. Vacca, Coord. Chem. Rev., 1999, 184, 311 CrossRef CAS.
- A. E. Martell and R. M. Smith, Standard Reference Database, 2001, vol. 46 Search PubMed.
- J. N. Weiss, FASEB J., 1997, 11, 835 CAS.
- F. C. Neidhardt, P. L. Bloch and D. F. Smith, J. Bacteriol., 1974, 119, 736 CAS.
- (a) S. L. Bloom and D. B. Zamble, Biochemistry, 2004, 43, 10029 CrossRef CAS; (b) P. T. Chivers and R. T. Sauer, Chem. Biol., 2002, 9, 1141 CrossRef CAS; (c) S. Leitch, M. J. Bradley, J. L. Rowe, P. T. Chivers and M. J. Maroney, J. Am. Chem. Soc., 2007, 129, 5085 CrossRef CAS.
- C. E. Outten and T. V. O'Halloran, Science, 2001, 292, 2488 CrossRef CAS.
- C. Dian, K. Schauer, U. Kapp, S. M. McSweeney, A. Labigne and L. Terradot, J. Mol. Biol., 2006, 361, 715 CrossRef CAS.
- B. Zambelli, M. Bellucci, A. Danielli, V. Scarlato and S. Ciurli, Chem. Commun., 2007, 43, 3649 Search PubMed.
- (a) L. O. Abraham, Y. Li and D. B. Zamble, J. Inorg. Biochem., 2006, 100, 1005 CrossRef CAS; (b) N. S. Dosanjh, N. A. Hammerbacher and S. L. Michel, Biochemistry, 2007, 46, 2520 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Materials and methods. Table S1 shows the CD fitting results, Table S2 gives ICP-MS quantification of metal ion loading to Hpn-FRET, and Table S3 shows the fitting results for binding curves of Hpn-FRET to Ni2+, Zn2+, and Bi3+, Fig. S1 shows protein gel of purified Hpn-FRET, Fig. S2 is the CD spectra of Hpn-FRET in the absence and presence of metals, Fig. S3 is the fluorescent spectra for the response of Zn2+, Co2+, and Ni2+ with Hpn-FRET, Fig. S4 displays the competition experiments for Zn2+ and Co2+ selectivity, Fig. S5 shows the fluorescence spectra of E. coli in the absence of additional metal, Fig. S6 shows the gel filtration chromatogram for purified Hpn-FRET, and Fig. S7 compares FRET response between Hpn-FRET and Hpn-FRET without a N-terminal His6-tag. See DOI: 10.1039/c0sc00411a |
| This journal is © The Royal Society of Chemistry 2011 |