Protecting the beneficial functionality of lipoproteins by 1-Fe, a corrole-based catalytic antioxidant†
Adi
Haber
a,
Michael
Aviram
*b and
Zeev
Gross
*a
aSchulich Faculty of Chemistry, Technion – Israel Institute of Technology, Technion City, Haifa, 32000, Israel. E-mail: chr10zg@tx.technion.ac.il
bLipid Research Laboratory, Rappaport Faculty of Medicine, Technion – Israel Institute of Technology, Rambam Medical Center, Haifa, 31096, Israel. E-mail: aviram@tx.technion.ac.il
First published on 29th October 2010
Abstract
Functionally defective HDL emerges as a main risk factor in cardiovascular diseases, on top of imbalanced serum lipoprotein ratios. We now show that 1-Fe, an iron(III) corrole catalytic antioxidant, spontaneously forms tightly-bound conjugates with LDL (secondarily) and HDL (primarily) of human serum. This is demonstrated by HPLC separation of serum/1-Fe mixtures and analysis of the corrole-containing fractions by UV-vis and circular dichroism spectroscopy. The LDL/1-Fe and HDL/1-Fe conjugates are resistant to functionality damaging events induced by oxidizing (copper salts) and nitrating species (peroxynitrite), as realized by heavily reduced formation of conjugated dienes, hydroperoxides, aldehydes, and nitrotyrosine. What is more, even already oxidized LDL becomes less atherogenic upon conjugation. Treatment with 1-Fe preserves the cardioprotective properties of HDL (cholesterol efflux, reduction of intracellular oxidative-stress, and inhibition of apoptosis), which are otherwise damaged under conditions of oxidative-stress. The novelty of 1-Fe relies in the unique bipolarity of the corrole macrocycle (responsible for its general affinity for lipoproteins), the affinity of the corrole-chelated iron(III) center to two histidine residues from (a particular subclass of) HDL-associated proteins, and the excellent catalytic activity regarding the decomposition of the most toxic naturally-occurring species/molecules.
Introduction
High ratio between the cholesterol-delivering and cholesterol-removing low- and high-density lipoproteins (LDL and HDL, respectively) is a well-recognized risk factor for cardiovascular diseases, which is traditionally treated by LDL-lowering medications or by HDL-increasing lifestyle.1 Oxidative stress, conditions characterized by an imbalance between the production of reactive oxygen/nitrogen species (ROS/RNS) and the system's ability to detoxify and remove them, has a major effect on lipoprotein quality. LDL and HDL particles modified by ROS and/or RNS (collectively termed oxLDL and oxHDL) are more atherogenic and less anti-atherogenic, respectively, than their native counterparts.2,3 The tremendous fundamental and practical research focus on LDL (the “bad cholesterol”) already led to highly significant breakthroughs. On the other hand, the consciousness of the need to investigate factors that may either induce or prevent the transformation of native HDL (the “good cholesterol”) into dysfunctional HDL is only nowadays sharply increasing.4,5The quite established knowledge regarding the function-damaging effects of ROS/RNS in numerous diseases led to an increasing awareness to the beneficial effects of antioxidant-rich diet,6 as well as to the popularity of appropriate food additives. The latter approach is, however, under critical review since several large-scale human surveys were quite disappointing.7 Plausible reasons for the apparent inconsistency between basic research and clinical trials include limited bioavailability of the supplied antioxidant molecules, their fast irreversible consumption, and their non-efficiency against some particular reactive species that damage the lipoproteins and the arterial wall.8 Hypothesis-driven research has strongly focused on the above two last mentioned aspects by developing metal-based antioxidants capable of decomposing the main ROS and RNS in a catalytic fashion.9 Some synthetic iron(III) and manganese(III) porphyrins are very potent catalytic antioxidants in terms of the rate that they decompose O2˙−,10HOONO (peroxynitrous acid, PN)11 and related species.12 Quite a variety of structurally optimized metalloporphyrins has indeed been successfully applied in models of the many diseases that are induced by oxidative stress.13–18 The importance of bioavailability has been highlighted by demonstrating that a slow acting lipophilic catalyst is as efficient in vivo as its more hydrophilic and much faster acting analog.19 Studies on the biodistribution of porphyrins and related macrocycles, mainly in the context of photodynamic therapy, have revealed very different serum distribution as a function of the number, location and kind of polar head groups.20–23
Metal complexes of the water soluble and bipolar corrole 1-H3 shown in Scheme 1 have recently been demonstrated to be of large utility in medicinal applications. The intensively fluorescent gallium(III) complex 1-Ga was used for following its uptake into breast cancer cells, which revealed that it is also capable of selective killing of those that over-express heregulin receptors (HER2-positive cells).24 This was followed by experiments that demonstrated both the imaging and the elimination of breast cancer tumors implanted in mice.25 These remarkable achievements were obtained when 1-Ga was conjugated to a protein that was originally devised for gene-delivery.
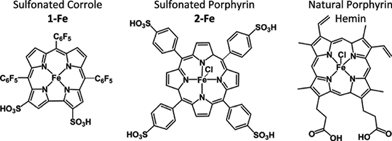 | ||
| Scheme 1 The chemical structures of compounds examined in this study. | ||
Parallel focus on the iron(III) and manganese(III) complexes of the same corrole (1-Fe and 1-Mn, respectively) was on molecular and cellular protection against oxidation and nitration, a potential that was disclosed in both chemical and biochemical systems. In particular, the iron(III) corrole 1-Fe displays excellent catalytic activity for decomposition of the main ROS and RNS that are involved in an exceedingly large variety of diseases: O2˙−,26H2O2,27 and HOONO.281-Mn and other water-soluble manganese(III) corroles exhibit slower catalytic rates, but they are highly advantageous for the attenuation of molecular and intracellular nitration.28 The relevance of these findings were identified in a variety of biochemical models relevant to diseases that are correlated with impaired ability of the natural systems to deal with excess ROS and RNS: diabetes,29 neurodegeneration,30 optic neuropathy,31 and atherosclerosis.32 The last mentioned investigation included also a mice-based model, in which 1-Fe attenuated the development of atherosclerotic lesions better than any natural or synthetic antioxidant reported so far.32 Importantly, no targeting protein was added in all the reported applications that rely on the iron(III) and manganese(III) corroles. Nevertheless, and in spite of the fact that derivatives with negatively charged sulfonato head groups are not expected to penetrate cell membranes, strong indications for cellular uptake were obtained by using the fluorescent gallium(III) analog 1-Ga. Independent investigations with isolated constitutes of serum revealed spontaneous and extremely strong association of this kind of metallocorroles to albumin33 and transferrin34 (Kd ≤ 1 and 10 nM, respectively), as well as to lipoproteins (about 40 corroles/LDL and 10 corroles/HDL).32 These findings suggest that serum proteins could be the carriers of corroles in the body and be involved in cellular uptake of these molecules. What is more, the natural functions of the proteins might be affected upon conjugation of the corrole.
The focus of the current investigations was on the elucidation of naturally present proteins in human serum as potential carriers of the most promising ROS/RNS decomposition catalyst, 1-Fe. We show that despite of the above mentioned very strong binding of this kind of corrole to isolated serum proteins, in whole serum there is an absolute preference for the cholesterol-carrying lipoproteins. Molecular level investigations of this unique phenomenon revealed that it is due to the combination of the specific bipolarity of the corrole and the coordination properties of the chelated iron(III). The latter aspect was found to be responsible for the different strength and kind of association of 1-Fe with distinctive classes and sub-classes of the lipoproteins. Some most vital functional aspects of the lipoproteins were affected by the spontaneous conjugation of the iron(III) corrole. The key findings are that 1-Fe decreased the pro-atherogenic effects of LDL (the “bad cholesterol”) and rescued the anti-atherogenic properties of HDL (the “good cholesterol”) from damage introduced by ROS and RNS. The novelty of these results may be appreciated by the increased awareness to the phenomenon of dysfunctional HDL even in populations that are not exposed to the traditional risk factors of cardiovascular disease.4
Results
Serum distribution of corroles
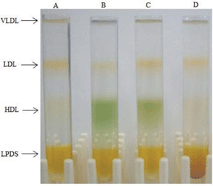 | ||
| Fig. 1 KBr density gradient separation of serum containing: A) no additive, B) 1-Fe, C) 1-Mn, and D) 2-Fe. | ||
These data (Fig. 2A and 2B) revealed, that 85% of 1-Fe was bound to HDL subfraction 2 (HDL2), whereas none was bound to HDL subfraction 3 (HDL3), and the remaining 15% was bound to LDL (the peak observed at the retention time of VLDL was due to other colored material, as it did not display the characteristic spectrum of 1-Fe). 1-Mn and 1-Ga displayed lower binding selectivity, with the corrole residing in all lipoprotein fractions. About 60% of these corroles was bound to HDL2, approximately 30% to LDL and the remaining distributed both to HDL3 and to VLDL (Fig. S1†).
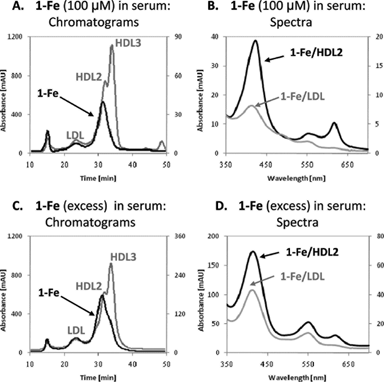 | ||
| Fig. 2 Serum distribution of 1-Fe. (A) The chromatograms at 280 nm (light line, relative to the left y-axis) and at 420 nm (dark line, relative to the right y-axis), obtained by HPLC separation of serum containing 100 μM 1-Fe. (B) The absorbance spectra of 1-Fe/HDL2 (dark line) and 1-Fe/LDL (light line), eluted under the conditions described in A. (C) The chromatograms at 280 nm (light line, relative to the left y-axis) and at 420 nm (dark line, relative to the right y-axis), obtained by HPLC separation of serum overloaded with 1-Fe and dialyzed for removing all loosely-bound or non-bound corrole. (D) The absorbance spectra of 1-Fe/HDL2 (dark line) and 1-Fe/LDL (light line), eluted under the conditions described in C. | ||
Similar HPLC experiments with 2-Fe revealed that this compound did not elute with any of the serum constitutes under the very diluting conditions of the HPLC experiment, whereas the natural porphyrin hemin (Scheme 1) was eluted within the HDL fraction (not shown).
Examining the 280 nm/420 nm absorption ratio at 31 min (corresponding to the HDL2 peak maxima) and comparing it to the ratio measured for solutions in which known amounts of 1-Fe were added to purified HDL2 allowed for determination of the molar ratio between 1-Fe and HDL within the conjugates. For the low 1-Fe concentration, one corrole molecule per two HDL2 particles was calculated, whereas two corrole molecules per one HDL2 particle were identified when using the high 1-Fe concentration.
Binding modes of corroles within lipoproteins
1-Mn conjugates with both HDL2 and LDL displayed practically identical absorbance spectra, which differed from that in buffered solutions with no additive by one main aspect. Upon addition of lipoproteins, the 480 nm band shifted to 476 nm and became the most intense absorbance peak (Fig. S2A and B†). For 1-Ga, no significant spectral change was detected following lipoprotein binding (Fig. S1D†).
![Mode of 1-Fe binding to lipoproteins. (A) Changes in absorbance spectra of 1-Fe upon its titration with histidine methyl ester, at 0, 25, 50 75, 100 and 125 [histidine]/[1-Fe] ratio. (B) Spectrum of 1-Fe with a very high histidine excess. (C) The visible circular dichroism spectra of 50 μM 1-Fe with 2.5 mg protein/mL HDL2 (dark line) or LDL (light line). (D) The absorbance spectrum of 1-Fe with apoE-containing liposomes.](/image/article/2011/SC/c0sc00448k/c0sc00448k-f3.gif) | ||
| Fig. 3 Mode of 1-Fe binding to lipoproteins. (A) Changes in absorbance spectra of 1-Fe upon its titration with histidine methyl ester, at 0, 25, 50 75, 100 and 125 [histidine]/[1-Fe] ratio. (B) Spectrum of 1-Fe with a very high histidine excess. (C) The visible circular dichroism spectra of 50 μM 1-Fe with 2.5 mg protein/mL HDL2 (dark line) or LDL (light line). (D) The absorbance spectrum of 1-Fe with apoE-containing liposomes. | ||
Titration of 1-Mn solutions with histidine displayed minor changes up to an extremely high histidine excess (>2000 fold, not shown).
Attenuation of oxidative damage to LDL and HDL
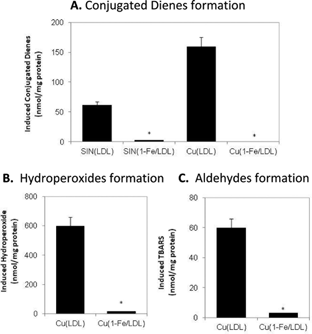 | ||
| Fig. 4 The effect of 1-Fe on LDL oxidative damage by SIN-1 or CuSO4. 0.1 mg protein/mL LDL was incubated with or without 2.5 μM 1-Fe for 10 min and then treated with 250 μM SIN-1 or 5 μM CuSO4 at 37 °C for 2 h. The amount of lipoprotein damage was determined by measuring formation of conjugated-dienes (A), hydroperoxides (B), and TBARS (C). | ||
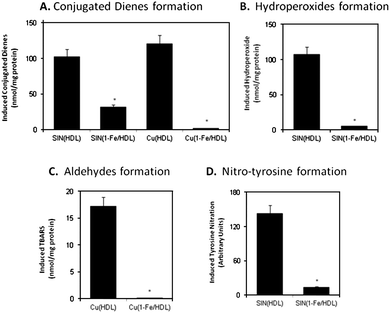 | ||
| Fig. 5 The effect of 1-Fe on HDL oxidative damage by SIN-1 or CuSO4. 1 mg protein/mL HDL was incubated with or without 50 μM 1-Fe for 10 min and then treated with 500 μM SIN-1 or 40 μM CuSO4 at 37 °C overnight. The amount of lipoprotein damage was determined by following the formation of conjugated-dienes (A), hydroperoxides (B), TBARS (C), and nitrated tyrosine (D). | ||
Atherogenicity of corrole-treated lipoproteins
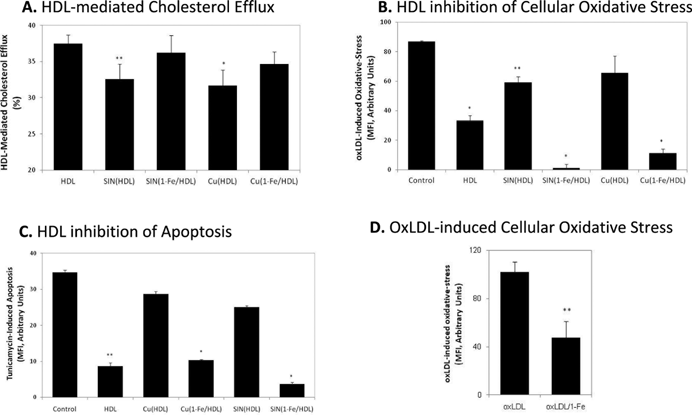 | ||
| Fig. 6 A–C: The effect of 1-Fe on oxidative-stress induced loss of anti-atherogenic HDL functions. 1 mg protein/mL HDL was incubated with or without 50 μM 1-Fe for 10 min and then treated with 500 μM SIN-1 or 40 μM CuSO4 at 37 °C overnight. Cholesterol efflux capacity (A), inhibition of oxLDL-induced oxidative-stress (measured by DCFH-DA) (B), and inhibition of tunicamycine-induced apoptosis (measured by DiOC6) (C) in J774 macrophages was determined as described in Materials and Methods. (D) Oxidative-stress induced in J774 macrophages with oxLDL pretreated overnight with or without 50 μM 1-Fe. Results are the mean of three independent experiments, ± SD. *P < 0.005, **P < 0.05 (vs. control). | ||
Discussion
Biodistribution of medications is a major factor dictating therapy efficacy, as demonstrated by many excellent in vitro performing drugs displaying limited in vivo efficiency.13–16,19 Every therapeutic that is administered into the body encounters the serum components, which may bind it and serve as carriers for its distribution between the various tissues. Accordingly, serum distribution of potential drugs is a main issue of their activity, and the first step in the disclosed research. We demonstrate an extremely strong association of the catalytic antioxidant 1-Fe to one particular sub-fraction of HDL (HDL2) and a somewhat lower affinity to LDL, on the expense of all other constituents of human serum. This spontaneous conjugation leads to significant reduction in oxidative/nitrative damage to both lipoproteins and, moreover, rescues biological activities of HDL that are crucial for its cardioprotective functions.The bis-sulfonated corrole investigated in the present research is bipolar, with negative charges on one side of the otherwise hydrophobic molecule, which explains its high binding affinity to the vehicles constructed for carrying the bipolar cholesterol in the bloodstream. This spontaneous association, which is strong enough as to withstand the very diluting chromatographic experiments, is apparently sufficient to outcompete the previously revealed very strong binding of this class of corroles to isolated serum proteins.33,34 This is not the case for the iron complex 2-Fe, a porphyrin-based PN decomposition catalyst with symmetrically distributed sulfonic acid head groups, which we have confirmed not to bind to any of the lipoproteins.35 The highly dissimilar distribution in serum may indicate a very different biological effect of this kind of corroles relative to analogous porphyrins, despite of similar catalytic activities. On the other hand, the bipolar hemin, with its two carboxylic acids located in a non-symmetric fashion, does bind to lipoproteins with high affinity. Hemin, however, has been established as a pro- rather than as an antioxidant.36
The different serum distribution pattern of 1-Fe, compared with those of 1-Mn and 1-Ga, points towards distinctive binding modes/motifs of the former in its interaction with lipoproteins. As these corroles differ only in the identity of the central metal, the chelated iron(III) ion is apparently responsible for the characteristics of 1-Fe association (vide infra). Regarding the other two corroles, similarity of spectral changes with both HDL2 and LDL signifies similar binding to both lipoprotein classes. The lack of CD signals for 1-Mn- and 1-Ga-lipoprotein conjugates indicates their association to non-chiral components therein. As liposomes induce the same spectral changes as do lipoproteins (whereas amino acids do not), it may be concluded that these two corroles are imbedded within the bipolar phospholipid layer present in all lipoproteins. Indeed, the higher corrole percentage in HDL2 relative to LDL and in LDL relative to VLDL may be easily explained on the basis of the relative molar concentrations of these particles within serum.37 Nevertheless, HDL3 molar concentration is at least as high as that of HDL2,4 and yet the lowest 1-Mn and 1-Ga binding percentage recorded is for HDL3. This may be attributed to the very low lipid content and surface area of HDL3, which are the lowest among all four lipoprotein (sub)classes, so that it probably just physically cannot bind many corrole molecules. The importance of lipid content and surface area for corrole binding may be appreciated by our previously published result regarding the maximal corrole-binding capacity of the lipoproteins, which in LDL is 4 times larger than in HDL.32
The first clue for understanding the highly selective binding of the iron(III) corrole to particular lipoproteins was provided by the differences in the absorbance spectra of 1-Fe/HDL2versus1-Fe/LDL, thus pointing towards different binding modes therein. What is more, serum saturated with 1-Fe displayed lower selectivity for HDL2 binding (with a distribution pattern more like that displayed by 1-Mn and 1-Ga), and the spectrum of the 1-Fe/HDL2 conjugated corrole was not identical to that obtained under limited corrole concentrations. In fact, the spectrum of 1-Fe/HDL2 from serum overloaded with corrole may be considered as a superposition of the spectra of 1-Fe/LDL conjugates and of 1-Fe/HDL2 conjugates under low corrole concentrations. This leads to the conclusion that HDL2 binds 1-Fe by different binding modes, of which the highest affinity one is specific to HDL2 (due to protein binding, vide infra) and the weaker one is general for lipoproteins (binding to the lipidic phase in the same manner as 1-Mn and 1-Ga). The second binding mode becomes apparent already at a corrole/HDL2 ratio of 2, indicative that there is only one super strong binding site per HDL2 particle.
Confirmation of the two different binding modes hypothesis, as well as additional insight regarding the nature of binding, was obtained by CD spectroscopy. This approach was adopted because of the significant differences between HDL and LDL in terms of their lipidversusprotein contents. LDL contains only 25 w/w% protein, composed of just one very large protein; apolipoprotein B100 (apoB100).37 In contrast, HDL particles possess a large variety of much smaller apolipoproteins (A's, E's, C's), as well as various enzymes [PON1, lecithin-cholesterol acyl-trasferase (LCAT), platelet-activating factor acyl-hydrolase (PAF-AH)], which for HDL2 account for about 40 w/w% of its content.4,38 Native LDL and HDL provide CD spectra with bands in the UV range due to their protein(s), but not in the visible. On the other hand, the visible light absorbing non-chiral corrole will display a CD spectrum only if located within a chiral environment. The visible CD spectrum obtained for 1-Fe/HDL2 solution, but not for 1-Fe/LDL solution, clearly points towards specific interactions of the corrole with proteins of HDL2, but not with apoB100 of LDL. The lack of CD signals for 1-Fe/LDL, as well as the analogous absorbance spectra observed for 1-Fe with LDL and with liposomes, further indicate that bis-sulfonated corroles bind to the lipid phase of LDL.
Independent evidence for 1-Fe binding to HDL-associated protein-derived moieties was obtained by examining the effect of basic amino acid esters, which may coordinate to the metal center of the corrole, on the electronic spectrum of 1-Fe in buffer. These experiments revealed that histidine, with its strongly coordinating imidazole side chain, induced spectral changes similar to those seen with HDL2, while all other relevant amino acids (Met, Tyr, Cys, Arg and Lys) did not affect the spectrum. Titration of 1-Fe with a histidine ester displayed isosbestic points only at low excess of the latter, consistent with the presence of three absorbing species at higher histidine excess: 1-Fe coordinated to water only, to one histidine, and to two histidine molecules. The absorbance spectrum (especially concerning the 620 nm band) was similar to that of 1-Fe/HDL2 conjugates only under conditions favoring the formation of bis-histidine-coordinated 1-Fe. Accordingly, it may be concluded that 1-Fe binds to HDL2 by interaction of the metal center with two closely positioned histidine residues from either one or two HDL2-associated apolipoproteins/enzymes. This may explain the high binding affinity and selectivity of 1-Fe to HDL2 at the expense of all other serum components. The much lower affinity of 1-Mn for histidine binding (demonstrated by the much higher histidine excess needed to induce spectral changes) may explain the lack of HDL2-selectivity for this corrole.
The quite low affinity of 1-Fe for bis-histidine coordination in the homogenous buffer solutionversus the high one in HDL2 is quite illuminating. It clearly suggests supramolecular arrangement of proteins in HDL2 that is perfectly suited for the interaction, a situation that apparently does not exist in HDL3. The excellent catalytic activity of HDL-conjugated corrole (vide infra), despite of the dominancy of the hexa-coordination of the metal therein, may be explained by fast equilibrium between the mono- and bis-histidine coordinated 1-Fe. A more detailed explanation for the discrimination between HDL2 and HDL3 by 1-Fe requires the identification of the specific protein(s) involved in the binding of the corrole, which is quite a formidable task because HDL particles contain numerous proteins and enzymes. The effect of two proteins on 1-Fe binding was nevertheless examined because of their quite different abundance in HDL sub-classes. Liposomes containing either apoE (resides mostly in HDL239) or PON1 (found mainly in HDL340) did not induce the formation of the 620 nm band that is characteristic of 1-Fe/HDL2 conjugates. Furthermore, the 620 nm band did not disappear in serum of knockout mice for either of these proteins. We must hence postpone elucidation of the protein(s) assembly responsible for 1-Fe binding to future investigation.
Possible impacts of all these findings were addressed by examining the utility of 1-Fe in protecting HDL and LDL against oxidative/nitrative damage, as well as on the consequential effects on their biological functions. HDL and 1-Fe/HDL conjugates, as well as LDL and 1-Fe/LDL conjugates, were treated with SIN-1, a reagent that provides a flux of PN at physiological pH.41 This particular damaging molecule was selected because: a) lipoprotein oxidation occurs chiefly in the arterial wall, where both precursors of PN (superoxide anion radical and nitric oxide) are produced;37 b) atherosclerotic plaques are rich in nitrated lipoproteins, a biomarker for PN involvement;16 c) 1-Fe is an excellent catalyst for PN decomposition;28 and d) the natural system manages PN extremely poorly. The examinations also included treatment with copper ions, the most commonly used model for inducing lipoprotein oxidation, which exert their damage through the formation of various ROS (including H2O2 and O2˙−). Reasons for the use of this oxidative system include the following: a) redox-active metal ions have been identified in human atherosclerotic plaques;42 b) LDL oxidized by copper ions displays specific oxidative markers that have been shown to be elevated in LDL from atherosclerotic lesions;43 and c) 1-Fe is an excellent catalyst for decomposition of both H2O2 and O2˙−.26,27
Lipid oxidation was evaluated by measuring formation of conjugated dienes (absorbance at 234 nm) and hydroperoxides (iodometric method) for SIN-1 treatment. For copper-induced oxidation the amount of aldehyde formation (thiobarbituric acid reactive substances, TBARS) was also measured, as transition metal ions efficiently catalyze the transformation of the hydroperoxide to the aldehydes. In both oxidation systems and for both lipoproteins examined, lipid oxidation was largely attenuated by 1-Fe. As SIN-1 is both a ROS and a RNS, proteintyrosine nitration was measured as well, and was found to be significantly reduced by pre-treatment of the lipoproteins with 1-Fe. These results show that the potent catalytic properties of 1-Fe are preserved upon conjugation to lipoproteins, i.e., the former prevents oxidative and nitrative damage of the latter.
The relevance of the above findings to possible effects of 1-Fe on the anti-atherogenic properties of HDL was demonstrated by focusing on three prominent functions of HDL: a) participation in cholesterol efflux from macrophages as to prevent their transformation to lipid-loaded foam cells, the hallmark of early atherosclerosis;44 b) reduction of oxidative stress in macrophages (and consequentially in arteries), which leads to lesion development;44 and c) cytoprotection of macrophages against apoptosis that leads to the formation of a necrotic core and may eventually cause plaque rapture.45 Consistent with expectation and literature reports, both copper and SIN-1 treatment of native HDL negatively affected these desirable biological activities. On the other hand, conjugation of 1-Fe to HDL completely eliminated the deleterious effect of these oxidants on all the above mentioned biological functions of HDL. We may thus safely conclude that the 1-Fe/HDL conjugate is much more resistant to oxidative stress induced loss of anti-atherogenic properties.
Interestingly, corrole-HDL conjugates (with or without further treatment with an oxidant) were more efficient than native HDL in reducing oxLDL-induced oxidative-stress in macrophages, indicating that the corrole exerts its antioxidative properties within this system as well. A critical examination of this hypothesis was performed by incubation of oxLDL with 1-Fe prior to its addition to the macrophages. In this case, the oxidative status of the cells was 35% lower relative to that with non-corrole-treated oxLDL. The possible roles of the corrole in this system are reduction of oxLDL oxidative status (such as by detoxification of the lipid hydroperoxides therein) or interference with the cellular oxidation process induced by oxLDL (by decomposition of secondary ROS produced by it). In any case, the outcome is that 1-Fe reduces the pro-atherogenic effect of oxLDL.
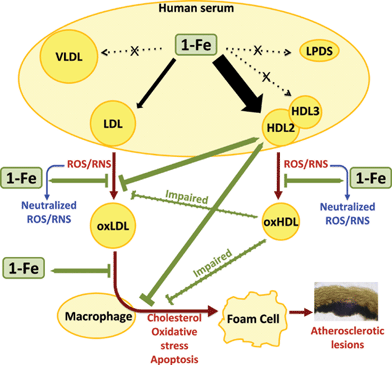 | ||
| Fig. 7 The selective affinity of 1-Fe to HDL2 and LDL of human serum and the consequential effects on their cardio protective functions. | ||
Summary and conclusions
The present research uncovers novel features of the iron(III) corrole 1-Fe, outlined in Fig. 7. The bipolarity of its corrole ligand is responsible for the general association to serum lipoproteins (a property shared with the analogous 1-Mn and 1-Ga), while the chelated iron(III) ion is involved in the binding to histidine residues of HDL2-associated protein(s), as well as in the catalytic decomposition of reactive oxygen and nitrogen species. 1-Fe binds spontaneously in a discriminatory fashion to HDL2 at the expense of all other human serum components except LDL, protects LDL and HDL against oxidative modifications and from the consequential loss of beneficial biological activities of HDL. In addition, it even reduces the disadvantageous pro-atherogenic effect of already oxidized LDL. The extremely strong binding of 1-Fe to HDL and LDL suggests that this catalytic ROS/RNS decomposition catalyst will be carried all the way to the arterial wall, where the need for its protective action against the atherogenicity-inducing damage to oxidized lipoprotein(s) is most crucial. This is not the case for the other frequently used ROS/RNS decomposition catalysts:13–18,46,47 since they commonly carry four symmetrically-distributed positively or negatively charged head groups, they are not strongly bound to lipoproteins.20–23 We expect the findings of the current work to be of prime importance for the design of optimal treatments for cardiovascular diseases, as many commonly used drugs and dietary antioxidants suffer from limited bioavailability, do not improve or rescue HDL's cardioprotective functions, or effectively reduce oxLDL pro-atherogenic actions.Acknowledgements
Financial support by The Israel Science Foundation (ZG) and The Herbert Irving Cancer and Atherosclerosis Research Fund (ZG) is highly acknowledged.Notes and references
- S. M. Grundy, J. I. Cleeman, C. N. B. Merz, H. B. Brewer, Jr, L. T. Clark, D. B. Hunninghake, R. C. Pasternak, S. C. Smith, Jr and N. J. Stone, Circulation, 2004, 110, 227–239 CrossRef.
- M. Aviram and M. Rosenblat, in Redox Genome Interactions in Health and Disease, ed. J. Fuchs, M. Podda and L. Packer, Marcel Dekker, NY, 2004, pp. 557–590 Search PubMed.
- M. Rosenblat, L. Gaidukov, O. Khersonsky, J. Vaya, R. Oren, D. S. Tawfik and M. Aviram, J. Biol. Chem., 2006, 281, 7657–7665 CrossRef CAS.
- A. Kontush and M. J. Chapman, Pharmacol. Rev., 2006, 58, 342–374 CrossRef CAS.
- J. D. Smith, Arterioscler., Thromb., Vasc. Biol., 2010, 30, 151–155 CrossRef CAS.
- L. J. Roberts, J. A. Oates, M. F. Linton, S. Fazio, B. P. Meador, M. D. Gross, Y. Shyr and J. D. Morrow, Free Radical Biol. Med., 2007, 43, 1388–1393 CrossRef CAS.
- G. Bjelakovic, D. Nikolova, L. L. Gluud, R. G. Simonetti and C. Gluud, JAMA, J. Am. Med. Assoc., 2007, 297, 842–857 CrossRef CAS.
- S. R. Steinhubl, Am. J. Cardiol., 2008, 101, 14D–19D CAS.
- B. J. Day, Drug Discovery Today, 2004, 9, 557–566 CrossRef CAS.
- G. DeFreitas-Silva, J. S. Reboucas, I. Spasojevic, L. Benov, Y. M. Idemori and I. Batinic-Haberle, Arch. Biochem. Biophys., 2008, 477, 105–112 CrossRef CAS.
- J. Lee, J. A. Hunt and J. T. Groves, J. Am. Chem. Soc., 1998, 120, 7493–7501 CrossRef CAS.
- M. Zdilla, A. Lee and M. Abu-Omar, Angew. Chem., Int. Ed., 2008, 47, 7697–7700 CrossRef CAS.
- L.-P. Liang, J. Huang, R. Fulton, B. J. Day and M. Patel, J. Neurosci., 2007, 27, 4326–4333 CrossRef CAS.
- T. Radovits, L. Seres, D. Gero, L.-n. Lin, C. J. Beller, S.-H. Chen, J. Zotkina, I. Berger, J. T. Groves, C. Szabo and G. Szabo, Mech. Ageing Dev., 2007, 128, 173–181 CrossRef CAS.
- A. S. Wu, M. Kiaei, N. Aguirre, J. P. Crow, N. Y. Calingasan, S. E. Browne and M. F. Beal, J. Neurochem., 2003, 85, 142–150 CrossRef CAS.
- C. Szabo, H. Ischiropoulos and R. Radi, Nat. Rev. Drug Discovery, 2007, 6, 662–680 CrossRef CAS.
- P. Pacher, L. Liaudet, P. Bai, J. G. Mabley, P. M. Kaminski, L. Virag, A. Deb, E. Szabo, Z. Ungvari, M. S. Wolin, J. T. Groves and C. Szabo, Circulation, 2003, 107, 896–904 CrossRef CAS.
- C. Szabó, J. G. Mabley, S. M. Moeller, R. Shimanovich, P. Pacher, L. Virág, F. G. Soriano, J. H. Van Duzer, W. Williams, A. L. Salzman and J. T. Groves, Mol. Med., 2002, 8, 571–580 CAS.
- I. Kos, L. Benov, I. Spasojevic, J. S. Reboucas and I. Batinic-Haberle, J. Med. Chem., 2009, 52, 7868–7872 CrossRef CAS.
- R. W. Boyle and D. Dolphin, Photochem. Photobiol., 1996, 64, 469–485 CrossRef CAS.
- K. Woodburn and D. Kessel, Int. J. Biochem. Cell Biol., 1995, 27, 499–506 CrossRef CAS.
- M. Kongshaug, J. Moan and S. B. Brown, Br. J. Cancer., 1989, 59, 184–188 CAS.
- D. Kessel, Cancer Lett., 1986, 33, 183–188 CrossRef CAS.
- H. Agadjanian, J. J. Weaver, A. Mahammed, A. Rentsendorj, S. Bass, J. Kim, I. J. Dmochowski, R. Margalit, H. B. Gray, Z. Gross and L. K. Medina-Kauwe, Pharm. Res., 2006, 23, 367–377 CrossRef CAS.
- H. Agadjanian, J. Ma, A. Rentsendorj, V. Valluripalli, J. Y. Hwang, A. Mahammed, D. L. Farkas, H. B. Gray, Z. Gross and L. K. Medina-Kauwe, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 6105–6110 CrossRef CAS.
- M. Eckshtain, I. Zilbermann, A. Mahammed, I. Saltsman, Z. Okun, E. Maimon, H. Cohen, D. Meyersteinc and Z. Gross, Dalton Trans., 2009, 7879–7882 RSC.
- A. Mahammed and Z. Gross, J. Am. Chem. Soc., 2005, 127, 2883–2887 CrossRef CAS.
- A. Mahammed and Z. Gross, Angew. Chem., Int. Ed., 2006, 45, 6544–6547 CrossRef CAS.
- Z. Okun, L. Kupershmidt, T. Amit, S. Mandel, O. Bar-Am, M. B. H. Youdim and Z. Gross, ACS Chem. Biol., 2009, 4, 910–914 CrossRef CAS.
- L. Kupershmidt, Z. Okun, T. Amit, S. Mandel, I. Saltsman, A. Mahammed, O. Bar-Am, Z. Gross and M. B. H. Youdim, J. Neurochem., 2010, 113, 363–373 CrossRef CAS.
- A. Kanamori, M. M. Catrinescu, A. Mahammed, Z. Gross and L. A. Levin, J. Neurochem., 2010, 114, 488–498 CAS.
- A. Haber, A. Mahammed, B. Fuhrman, N. Volkova, R. Coleman, T. Hayek, M. Aviram and Z. Gross, Angew. Chem., Int. Ed., 2008, 47, 7896–7900 CrossRef CAS.
- A. Mahammed, H. B. Gray, J. J. Weaver, K. Sorasaenee and Z. Gross, Bioconjugate Chem., 2004, 15, 738–746 CrossRef CAS.
- A. Haber, H. Agadjanian, L. K. Medina-Kauwe and Z. Gross, J. Inorg. Biochem., 2008, 102, 446–457 CrossRef CAS.
- P. C. De Smidt, A. J. Versluis and T. J. C. Van Berkel, Biochem., 2002, 32, 2916–2922.
- G. Camejo, C. Halberg, A. Manschik-Lundin, E. Hurt-Camejo, B. Rosengren, H. Olsson, G. I. Hansson, G. B. Forsberg and B. Ylhen, J. Lipid Res., 1998, 39, 755–766 CAS.
- Oxidative stress, lipoproteins and cardiovascular dysfunction, ed. C. Rice-Evans and K. R. Bruckdorfer, Portland Press, London, 1995 Search PubMed.
- V. Zannis, A. Chroni and M. Krieger, J. Mol. Med., 2006, 84, 276–294 CrossRef CAS.
- M. C. Cheung and J. J. Albers, J. Lipid Res., 1982, 23, 747–753 CAS.
- C. Bergmeier, R. Siekmeier and W. Gross, Clin. Chem., 2004, 50, 2309–2315 CrossRef CAS.
- F. J. Martin-Romero, Y. Gutiérrez-Martin, F. Henao and C. Gutiérrez-Merino, J. Fluoresc., 2004, 14, 17–23 CrossRef CAS.
- N. Stadler, R. A. Lindner and M. J. Davies, Arterioscler., Thromb., Vasc. Biol., 2004, 24, 949–954 CrossRef CAS.
- C. Leeuwenburgh, J. E. Rasmussen, F. F. Hsu, D. M. Mueller, S. Pennathur and J. W. Heinecke, J. Biol. Chem., 1997, 272, 3520–3526 CrossRef CAS.
- M. Rosenblat, R. Karry and M. Aviram, Atherosclerosis, 2006, 187, 74–81 CAS.
- B. Fuhrman, A. Gantman and M. Aviram, Atherosclerosis, 2010, 211, 61–68 CrossRef CAS.
- A. Trostchansky, G. Ferrer-Sueta, C. Batthyany, H. Botti, I. Batinic-Haberle, R. Radi and H. Rubbo, Free Radical Biol. Med., 2003, 35, 1293–1300 CrossRef CAS.
- A. Bloodsworth, V. B. O'Donnell, I. Batinic-Haberle, P. H. Chumley, J. B. Hurt, B. J. Day, J. P. Crow and B. A. Freeman, Free Radical Biol. Med., 2000, 28, 1017–1029 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental section and figures S1 and S2. See DOI: 10.1039/c0sc00448k |
| This journal is © The Royal Society of Chemistry 2011 |