Carbon monoxide coupling and functionalisation at a simple uranium coordination complex†
Polly L.
Arnold
*a,
Zoë R.
Turner
a,
Ronan M.
Bellabarba
b and
Robert P.
Tooze
b
aEaStCHEM School of Chemistry, Joseph Black Building, University of Edinburgh, Edinburgh, UK EH9 3JJ. E-mail: Polly.Arnold@ed.ac.uk
bSasol Technology UK, Purdie Building, North Haugh, St Andrews, UK KY16 9SR
First published on 28th October 2010
Abstract
A simple coordination complex of uranium(III), a uranium tris(amide), can selectively couple gaseous CO to the linear ynediolate [OCCO]2− dianion, at room temperature and pressure, regardless of the reagent stoichiometry. This product exhibits further reactivity upon warming in the form of the addition of a C–H bond of a methyl group across the C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C triple bond, this second carbon–carbon bond forming reaction generating a functionalised enediolate dianion.
C triple bond, this second carbon–carbon bond forming reaction generating a functionalised enediolate dianion.
Introduction
Carbon monoxide is the key C1 component in the Fischer–Tropsch process and the chemical industry worldwide. It is generated from biomass or coal in the form of synthesis gas, or syngas, (CO + nH2) and used to make millions of tonnes of solvents, fuels, and bulk chemicals every year. The bond in CO is the strongest in the periodic table, and whilst many metal complexes are capable of catalysing the insertion of CO into other organic substrates, only a handful of complexes have demonstrated the C–C coupling of two metal-coordinated CO ligands. Molten potassium was reported to reductively couple CO to form salts of the oxocarbon anions from homologues to [C2O2]2− up to [C5O5]2− and [C6O6]2− in the early nineteenth century.1,2 The reduction of Ta(CO)2(dmpe)2Cl by magnesium in the presence of Cp2ZrCl2 affords coupled and silylated Ta(Me3SiOCCOSiMe3)(dmpe)2Cl (dmpe = 1,2-bis{dimethylphosphino}ethane)3 and a study on the insertion of one and two equivalents of CO into the weak Rh–Rh bond in the rhodium octaethylporphyrin (oep) dimer [Rh(oep)]2 used 12 atm pressures of CO to force an equilibrium reaction over to a double insertion product characterised by 1H NMR spectroscopy as (oep)Rh(CO)(CO)Rh(oep).4 A heterotrimetallic tantalum hydride was shown to reductively couple six CO molecules at ambient pressure and temperature; protonolysis with [Me3NH][BPh4] yielded [Ta(2,6-(CH2-3-tBu-5-Me-2-OPh)(thf)]4(C6O6).5 Reactions that can form carbon–carbon bonds are rare in general, and the direct dimerisation of CO to afford OCCO is energetically uphill by +73 kcal mol−1, and particularly rare.1 Though the insertion of CO into metal alkyl, amido and hydride bonds is now well documented for Group 4 and f-block metal complexes,6–14 there are only two complexes which bind and reductively couple carbon monoxide to date; the sterically congested, trivalent, organometallic uranium complexes [U(η-C8H6{SiiPr3-1,4}2)(η5-CpR)] (CpR = C5(CH3)5 or C5(CH3)4H).15–17 The former demonstrated unprecedented reductive trimerisation of CO. The same UIII complex is also capable of reductively dimerising CO if exactly one equivalent of CO gas is used, although no interconversion between the (CO)n homologues, or subsequent reactivity, has been demonstrated to date. Reductive homologation by the strongly reducing LaIII and SmII complexes, [La(η-Cp*)2]2(N2) and [Sm(η-Cp*)2(thf)2], affords a ketene carboxylate dianion from three CO molecules at 90 psi pressures in 19 and 20% isolated yield, respectively.18,19 The incorporation of a pendant borane group to provide a hydride source has recently been used to address the second challenge in CO coupling – the formation of the reduced C–C and C–H bonds, the carbonyl [(Ph2P(CH2)2B(C8H14))2Re(CO)4][BF4] is borane activated to form Re–CH2–O–BR3 which couples with a second carbonyl equivalent.20 As yet, no catalytic C–C coupling or oligomerisation reactions exist for carbon monoxide at low pressures or temperatures.21
The tris(amido) complex of uranium UN″3, where N″ is the hydrocarbon-soluble, sterically demanding, bis(alkylsilyl)amido monoanion N(SiMe3)2−, is widely used as a reagent in coordination chemistry, and despite its pyramidal structure, it has generally been regarded not to possess any interesting small molecule activation chemistry of its own. The 1979 report of its synthesis reported the authors' surprise at the absence of reactivity with CO or similar Lewis bases such as tetrahydrofuran (THF), phosphines or isonitriles.22 Since then, the 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 adduct with the strong nucleophilic donor N-heterocyclic carbene tetramethylimidazol-2-ylidene (NHC),23 and with a further equivalent of the potassium amide KN″ have been reported;24 the products (NHC)UN″3 and K[UN″4], respectively, demonstrating that there is sufficient space at the metal centre for further reaction chemistry.
:1 adduct with the strong nucleophilic donor N-heterocyclic carbene tetramethylimidazol-2-ylidene (NHC),23 and with a further equivalent of the potassium amide KN″ have been reported;24 the products (NHC)UN″3 and K[UN″4], respectively, demonstrating that there is sufficient space at the metal centre for further reaction chemistry.
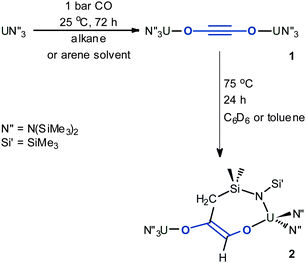
Herein, we report how hydrocarbon solutions of the simple trivalent uranium amide UN″3 react at ambient temperatures and pressures with carbon monoxide to afford a reductively coupled ynediolate [OCCO]2− unit, and how, for the first time, further reactivity is demonstrated for the linear ynediolate fragment. The simplicity, selectivity and redox-reactivity of this system suggest interesting catalytic carbon monoxide functionalisation chemistry may soon be accessible with related uranium(III) salts.
Results and discussion
Exposure of a dark purple hydrocarbon solution of the uranium amide complex UN″3 (N″ = N(SiMe3)2) to carbon monoxide, CO, at ambient temperature and pressure results in a gradual decolourisation of the solution to amber. Golden-coloured crystals of the UIV complex N″3U–OCCO–UN″31 were formed in 82% yield, as the product of a reductive coupling of CO between two U centres, Scheme 1.‡ We note that the original report of the inertness of UN″3 towards CO did not include details of solvent;22 the formation of 1 works equally well in hexanes or toluene, but not in the donor solvents tetrahydrofuran (THF) or pyridine. The 1H NMR spectrum of a benzene solution of 1 contains a single paramagnetically shifted resonance for the silylmethyl H nuclei at −8.6 ppm (parts per million). The reaction can be carried out readily on a gram scale, and if repeated with 13C-carbon monoxide, a resonance at 171 ppm is observed in the 13C NMR spectrum for the ynediolate carbon nuclei in N″3U–O13C13CO–UN″31a. The carbon resonance was not visible in the absence of 13C labelling. A single-crystal X-ray diffraction experiment confirms the one-electron oxidation of the uranium to UIV, which we assume drives the reaction, and the linearity of the UOCCOU unit in 1 (U–O–C = 178°); Fig. 1a shows the core of the molecular structure of 1. The formerly pyramidal geometry at the uranium centre has flattened out with the U cation dropping from 0.46 Å above the plane described by the three N atoms (in UN″3)25 to 0.16 Å (in 1) and the average U–N bond distance has shortened from 2.320 Å (in UN″3) to 2.253 Å (in 1), in accordance with the oxidation of the metal to UIV.
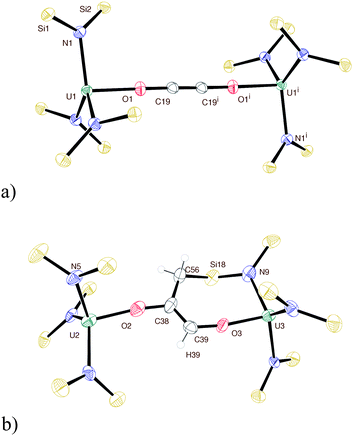 | ||
| Fig. 1 Displacement ellipsoid drawing of the molecular structure of (a) 1 and (b) 2 (50% probability) with all silyl methyl groups omitted for clarity. Selected distances (Å) and angles (°): 1: U1–O1 2.102(2), O1–C19 1.301(4), C19-C19i 1.183(7), U1–N1 2.251(3), O1–U1–N1 95.12(10), N1–U1–N2 122.01(10); 2: U2–O2 2.093(7), O2–C38 1.402(11), C38–C39 1.310(13), U2–N5 2.242(7), U2–O2–C38 171.6(6), U3–O3–C39 170.9(6), O2–U2–N5 98.9(3), C56–C38–C39 125.8(9), N1–U1–N2 116.3(3). | ||
 | ||
| Scheme 1 Coupling and functionalisation of carbon monoxide by the trivalent uranium amide to form a uranium-coordinated ynediolate 1, and then an ene-diolate 2. | ||
The first example of CO coordination to uranium was observed in 1986; tris(cyclopentadienyl)UIII was shown to bind CO reversibly,26 with substituted cyclopentadienyl and organometallic analogues later demonstrated to show stronger binding.27–29 The first non-organometallic uranium carbonyl complex was formed by reductive activation of a single CO molecule by two UIIIL complexes, in which L is a bulky tris(aryloxide) ligand, forming [LU3.5]2(μ:η1,η1-CO).30 No further reactivity of these interesting complexes has yet been reported. In the sole previous example of the reductive coupling of carbon monoxide to the coordinated ynediolate no further reaction was observed; a result corroborated by computational analysis.16 In contrast, heating a sample of 1 in benzene results in the formation of asymmetric 2, Scheme 1, in which one of the amido Si–CH3 groups has reacted formally to add C–H across the alkyne group intramolecularly, affording a seven-membered metallacyclic ring containing the newly formed alkene. Identified by 1H NMR spectroscopy (containing six paramagnetically shifted resonances), and a single-crystal X-ray diffraction study, Fig. 1b, the gold-coloured complex still contains two UIV cations, with close to linear U–O–C moieties, a planar O–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C–O group, and an average UIV–N distance of 2.266 Å.
C–O group, and an average UIV–N distance of 2.266 Å.
Both the reactions to form 1 and 2 with 13C-labelled carbon monoxide were monitored by in situ NMR spectroscopies, but no evidence of any intermediates was observed in either case. Further monitoring was hampered by the fact that both products tend to crystallise out of the solution as they form, and the range of solvents in which the formation of 1 can be monitored is limited to very non-polar, non-coordinating solvents.
Further chemistry of both 1 and 2 has also been investigated, to identify the potential for intermolecular functionalisation or substitution of the coupled oxycarbon anion. No reaction was observed between either 1 or 2 and dihydrogen at 1 atm pressure in benzene or THF solvent. The exposure of solutions of UN″3 and the cholorosilane ClSiMe3 to an atmosphere of CO result in the formation of coupled 1 before any other appreciable reaction occurs, and warming the mixture still leads to the formation of 2 in preference to any other reactivity, presumably due to the obvious steric crowding in the complex. No reaction between 1 and other silanes, boranes and haloboranes (see ESI†) was observed, and heating of these reaction mixtures again afforded only 2.
Conclusions
Hydrocarbon solutions of the simple trivalent uranium amide UN″3 react at ambient temperatures and pressures with carbon monoxide to afford a reductively coupled ynediolate [OCCO]2− unit. The silylamido ligands are too sterically encumbering to allow any intermolecular reaction to occur in this system with reagents tried so far, but the electronic structure of this tris(amido) uranium system is clearly able to support further reactivity of the ynediolate fragment, exemplified here by the second carbon–carbon bond and carbon–hydrogen bond forming reactions occurring. To our knowledge, this is the first time that a metal complex has been able to reductively couple exclusively two molecules of gaseous CO at atmospheric pressure, and the first time that this reductively coupled (CO)2 has been demonstrated to form bonds to sp3 carbon and hydrogen atoms.An increasing range of reductive activation reactions of small, traditionally inert molecules such as dinitrogen and carbon dioxide are being demonstrated for trivalent uranium complexes,31–35 and the ready interconversion between the UIII and UIV oxidation states involved here suggests that catalytic systems based on this coupling and functionalisation are viable. It is notable that the reaction occurs with such a simple coordination compound – an amide that is made from simple commercially available ligands (the precursor amide salt currently costs under €100 per mol). Work is in progress to develop mild routes to remove the coupled diolate with other reagents, and complete such a cycle.
Acknowledgements
We thank Dr Stephen M. Mansell for collection of some of the analytical data, and Sasol Technology UK (studentship for Z. R. T.), the UK EPSRC (fellowship for P. L. A.) and the University of Edinburgh for funding.Notes and references
- B. B. Wayland and X. Fu, Science, 2006, 311, 790 CrossRef CAS.
- W. Büchner, Helv. Chim. Acta, 1963, 46, 2111.
- P. A. Bianconi, I. D. Williams, M. P. Engeler and S. J. Lippard, J. Am. Chem. Soc., 1986, 108, 311 CrossRef CAS.
- V. L. Coffin, W. Brennen and B. B. Wayland, J. Am. Chem. Soc., 1988, 110, 6063 CrossRef CAS.
- T. Watanabe, Y. Ishida, T. Matsuo and H. Kawaguchi, J. Am. Chem. Soc., 2009, 131, 3474 CrossRef CAS.
- P. T. Wolczanski and J. E. Bercaw, Acc. Chem. Res., 1980, 13, 121 CrossRef.
- T. J. Marks, Science, 1982, 217, 989 CAS.
- W. J. Evans, J. W. Grate and R. J. Doedens, J. Am. Chem. Soc., 1985, 107, 1671 CrossRef CAS.
- K. G. Moloy, P. J. Fagan, J. M. Manriquez and T. J. Marks, J. Am. Chem. Soc., 1986, 108, 56 CrossRef CAS.
- K. G. Moloy and T. J. Marks, J. Am. Chem. Soc., 1984, 106, 7051 CrossRef CAS.
- K. Tatsumi, A. Nakamura, P. Hofmann, R. Hoffmann, K. G. Moloy and T. J. Marks, J. Am. Chem. Soc., 1986, 108, 4467 CrossRef CAS.
- W. J. Evans, S. A. Kozimor, G. W. Nyce and J. W. Ziller, J. Am. Chem. Soc., 2003, 125, 13831 CrossRef CAS.
- E. L. Werkema, L. Maron, O. Eisenstein and R. A. Andersen, J. Am. Chem. Soc., 2007, 129, 6662 CrossRef CAS.
- O. Bénaud, J. C. Berthet, P. Thuéry and M. Ephritikhine, Inorg. Chem., 2010, 49, 8117 CrossRef CAS.
- O. T. Summerscales, F. G. N. Cloke, P. B. Hitchcock, J. C. Green and N. Hazari, J. Am. Chem. Soc., 2006, 128, 9602 CrossRef CAS.
- O. T. Summerscales, F. G. N. Cloke, P. B. Hitchcock, J. C. Green and N. Hazari, Science, 2006, 311, 829 CrossRef.
- A. S. Frey, F. G. N. Cloke, P. B. Hitchcock, I. J. Day, J. C. Green and G. Aitken, J. Am. Chem. Soc., 2008, 130, 13816 CrossRef CAS.
- W. J. Evans, J. W. Grate, L. A. Hughes, H. Zhang and J. L. Atwood, J. Am. Chem. Soc., 1985, 107, 3728 CrossRef CAS.
- W. J. Evans, D. S. Lee, J. W. Ziller and N. Kaltsoyannis, J. Am. Chem. Soc., 2006, 128, 14176 CrossRef CAS.
- A. J. M. Miller, J. A. Labinger and J. E. Bercaw, J. Am. Chem. Soc., 2008, 130, 11874 CrossRef CAS.
- E. M. Carnahan, J. D. Protasiewicz and S. J. Lippard, Acc. Chem. Res., 1993, 26, 90 CrossRef CAS.
- R. A. Andersen, Inorg. Chem., 1979, 18, 1507 CrossRef CAS.
- H. Nakai, X. L. Hu, L. N. Zakharov, A. L. Rheingold and K. Meyer, Inorg. Chem., 2004, 43, 855 CrossRef CAS.
- W. J. Evans, D. S. Lee, D. B. Rego, J. M. Perotti, S. A. Kozimor, E. K. Moore and J. W. Ziller, J. Am. Chem. Soc., 2004, 126, 14574 CrossRef CAS.
- J. L. Stewart and R. A. Andersen, Polyhedron, 1998, 17, 953 CrossRef CAS.
- J. G. Brennan, R. A. Andersen and J. L. Robbins, J. Am. Chem. Soc., 1986, 108, 335 CrossRef CAS.
- M. D. Conejo, J. S. Parry, E. Carmona, M. Schultz, J. G. Brennan, S. M. Beshouri, R. A. Andersen, R. D. Rogers, S. Coles and M. Hursthouse, Chem.–Eur. J., 1999, 5, 3000 CrossRef CAS.
- J. Parry, E. Carmona, S. Coles and M. Hursthouse, J. Am. Chem. Soc., 1995, 117, 2649 CrossRef CAS.
- W. J. Evans, S. A. Kozimor, G. W. Nyce and J. W. Ziller, J. Am. Chem. Soc., 2003, 125, 13831 CrossRef CAS.
- I. Castro-Rodriguez and K. Meyer, J. Am. Chem. Soc., 2005, 127, 11242 CrossRef CAS.
- A. R. Fox, S. C. Bart, K. Meyer and C. C. Cummins, Nature, 2008, 455, 341 CrossRef CAS.
- O. T. Summerscales and F. G. N. Cloke, Struct. Bonding, 2008, 127, 87.
- I. Korobkov, S. Gorelsky and S. Gambarotta, J. Am. Chem. Soc., 2009, 131, 10406 CrossRef CAS.
- M. Ephritikhine, Dalton Trans., 2006, 2501 RSC.
- I. Castro-Rodriguez, H. Nakai, L. N. Zakharov, A. L. Rheingold and K. Meyer, Science, 2004, 305, 1757 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental and crystallographic details (CCDC reference numbers 775504 and 775505) and spectroscopic data. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c0sc00452a |
| ‡ To a freeze-pump-thaw degassed solution of UN″3 (1.00 g, 1.39 mmol) in toluene (20 mL) was added CO (1 atm) at room temperature. After 1 h, golden coloured microcrystalline solid started to precipitate and the reaction mixture was then kept at room temperature for 3 days. The solid was isolated by filtration, washed with hexane (3 × 10 mL) and then dried in vacuo to afford N″3UOCCOUN″31 as a golden-coloured crystalline solid. Yield 0.853 g (82%). Anal. (%): found (calc. for C38H108N6O2Si12U2): C, 30.37 (30.54); H, 7.35(7.28); N, 5.55 (5.62). 1H NMR (C6D6, 599.8 MHz, 298 K): δ 8.55 (108 H, s, N{Si(CH3)3}2) (fwhm = 1297 Hz) ppm. No ynediolate stretch was visible in the FTIR spectrum. Heating a slurry of N″3UOCCOUN″31 (0.107 g, 0.0716 mmol) in toluene (10 mL) to 80 °C for 24 h resulted in the conversion to N″3UOCC(H)OU{N(SiMe2CH2)(SiMe3)}N″22, also a golden-coloured solid, in 0.0713 g (67%) yield. Single crystals suitable for X-ray structure determinations were grown from room temperature benzene and toluene solutions of 1 and 2, respectively. |
| This journal is © The Royal Society of Chemistry 2011 |