Synthesis and carbene-transfer reactivity of dimeric nickel carbene cations supported by N-heterocyclic carbene ligands†
Carl A.
Laskowski
and
Gregory L.
Hillhouse
*
Gordon Center for Integrative Science, Department of Chemistry, The University of Chicago, 929 E. 57th St., Chicago, IL 60637, USA. E-mail: g-hillhouse@uchicago.edu; Fax: +773-702-0805; Tel: +773-702-7057
First published on 3rd November 2010
Abstract
Dimeric nickel bridging-carbene complex salts [{(IPr)Ni}2(μ-Cl)(μ-CR1R2)][B(ArF)4](6, R1 = R2 = Ph; 7, R1 = H, R2 = SiMe3) are synthesized by reaction of {(IPr)Ni(μ-Cl)}2 (3) with NaB(ArF)4 and N2CPh2 or N2CHSiMe3, with elimination of N2 and NaCl. The solid-state structure of 6 features an unsymmetric μ,η3-bonding motif for the bridging diphenylcarbene ligand in which one Ni center binds to the CPh2 ligand in a π-fashion involving the ipso- and one ortho-carbon of a phenyl ring along with the carbene carbon. The solution structure (NMR) of 6 indicates a C2v-symmetric structure even at −80 °C. The solid-state structure of 7 shows a (trimethylsilyl)carbene unit symmetrically disposed between the two nickel centers and bound through only the carbene-carbon atom. Diphenylcarbene-group transfer from 6 to carbon monoxide (3 equiv) gives O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CCPh2 and the dimeric Ni(I)-Ni(I) carbonyl complex [{(IPr)Ni(CO)}2(μ-Cl)][B(ArF)4] (8). Excess pivaloisocyanide reacts with 6 to afford tBuNCCPh2, [(IPr)Ni(CNtBu)3][B(ArF)4] (9), and (IPr)NiCl(CNtBu) (10). Mesitylazide reacts with 6 to give the ketimine MesNCPh2 and the bridging mesitylimido dimer [{(IPr)Ni}2(μ-Cl)(μ-NMes)][B(ArF)4] (5). A secondary reaction of diphenyldiazomethane with 6 to give Ph2CCPh2 and Ph2CN–NCPh2 prevents catalytic CPh2-group transfer reactions from being realized in these systems.
CCPh2 and the dimeric Ni(I)-Ni(I) carbonyl complex [{(IPr)Ni(CO)}2(μ-Cl)][B(ArF)4] (8). Excess pivaloisocyanide reacts with 6 to afford tBuNCCPh2, [(IPr)Ni(CNtBu)3][B(ArF)4] (9), and (IPr)NiCl(CNtBu) (10). Mesitylazide reacts with 6 to give the ketimine MesNCPh2 and the bridging mesitylimido dimer [{(IPr)Ni}2(μ-Cl)(μ-NMes)][B(ArF)4] (5). A secondary reaction of diphenyldiazomethane with 6 to give Ph2CCPh2 and Ph2CN–NCPh2 prevents catalytic CPh2-group transfer reactions from being realized in these systems.
Introduction
Transition metal carbene complexes are important reagents and intermediates in many synthetically useful reactions, including olefin metathesis and cyclopropanation.1 We have reported the isolation and characterization of a monomeric nickel carbene, (dtbpe)NiCPh2 (1, dtbpe = 1,2-bis(di-tert-butylphosphino)ethane), from (dtbpe)Ni(cod) and diphenyl–diazomethane (N2CPh2) (cod = 1,5-cyclooctadiene; Scheme 1).2Group transfer reactions from 1 to form cyclopropane, cyclopropene, ketimine, ketene, and ketone products have been demonstrated.3,4 However, carbene-transfer catalysis with the dtbpe system is very limited (a few turnovers for the cyclopropanation of 1-hexene) due to azo-coupling between 1 and diphenyldiazomethane to form tetraphenylazine, Ph2CN–NCPh2.3b In related chemistry, imido complexes (dtbpe)NiNR (2, Scheme 1) can be prepared from the appropriate organoazide (N3R) and {(dtbpe)Ni}2(C6H6), and undergo similar stoichiometric “nitrene” transfer reactions with appropriate substrates to give aziridine, carbodiimide, isocyanate, and diazene derivatives.3a,4Catalytic nitrene transfer from 2 is also limited by the reaction of 2 with N3R to give diazenes (RNNR) and dinitrogen at faster rates than for nitrene-transfer to other substrates.4
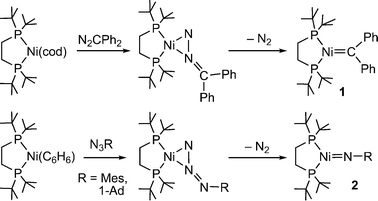 | ||
| Scheme 1 Synthesis of 1 from N2CPh2 and (dtbpe)Ni(cod) with N2 elimination. Related imido complexes (2) can be prepared from organoazides. Mes = 2,4,6-trimethylphenyl, 1-Ad = 1-adamantyl. | ||
The undesirable reaction between nickel-imido moieties and N3R noted above can be obviated by using bimetallic frameworks containing N-heterocyclic carbene ancillary ligands.5 We have shown that the Ni(I)-Ni(I) dimer {(IPr)Ni(μ-Cl)}2 (3, IPr = 1,3-(2,6-iPr2C6H3)2imidazolin-2-ylidene) reacts with excess N3Mes to form the Ni(II)-Ni(II) bridging imido complex {(IPr)NiCl}2(μ-NMes) (4).5 Removal of a chloride ligand from 4 gives the complex salt [{(IPr)Ni}2(μ-Cl)(μ-NMes)][B(ArF)4] (5; B(ArF)4 = B{3,5-(CF3)2C6H3)}4; Scheme 2). Importantly, neither 4 nor 5 react further with N3Mes, and are competent catalysts for the formation of carbodiimides (MesNCNR) and mesityl isocyanate (MesNCO) from N3Mes and isocyanides or carbon monoxide, respectively (illustrated for carbodiimide synthesis in Scheme 2).
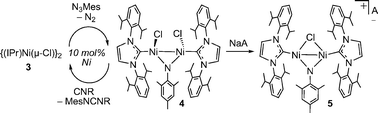 | ||
| Scheme 2 Catalytic carbodiimide synthesis with a NHC-supported dimer 4. Catalytic mesityl isocyanate formation can be accomplished with 5. A− = B(ArF)4− = B{3,5-(CF3)2C6H3)}4−. (See ref. 5) | ||
We were intrigued by the possibility that a similar synthetic strategy using 3 and diazoalkanes might afford access to dimeric complexes with bridging carbene ligands that could participate in group-transfer reactions. Herein we describe the synthesis and characterization of two bridging dinickel carbene complexes from 3 with diazoalkanes serving as the carbene synthons. The reactivity of the new carbene complexes and their potential catalytic efficacy in group-transfer reactions are evaluated and compared with the structurally analogous bridging-imido complexes.
Results and discussion
Carbene syntheses and structures
In an attempt to access a carbene analogue of the bridging imido complex 4, we investigated the reaction of the Ni(I) chloride dimer 3 with diphenyldiazomethane. Stoichiometric (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) addition of N2CPh2 to diethyl ether solutions of 3 resulted in vigorous evolution of N2 and darkening of the solution. A 1H-NMR spectrum of the crude reaction mixture showed formation of multiple products (along with unreacted 3; see discussion below), and while resonances consistent with a product of the formulation “{(IPr)Ni(Cl)}2(μ-CPh2)” were observed, attempts to isolate the neutral carbene dimer by selective crystallization were unsuccessful. In contrast, addition of one equivalent of N2CPh2 to a stirred diethyl ether solution containing equimolar amounts of 3 and NaB(ArF)4 resulted in N2 evolution and clean formation of the diphenylcarbene complex salt [{(IPr)Ni}2(μ-Cl)(μ-CPh2)][B(ArF)4] (6) as forest-green crystals in 89% isolated yield (Scheme 3). Chloride abstraction with NaB(ArF)4 facilitated removal of neutral contaminants in a simple manner.
:1) addition of N2CPh2 to diethyl ether solutions of 3 resulted in vigorous evolution of N2 and darkening of the solution. A 1H-NMR spectrum of the crude reaction mixture showed formation of multiple products (along with unreacted 3; see discussion below), and while resonances consistent with a product of the formulation “{(IPr)Ni(Cl)}2(μ-CPh2)” were observed, attempts to isolate the neutral carbene dimer by selective crystallization were unsuccessful. In contrast, addition of one equivalent of N2CPh2 to a stirred diethyl ether solution containing equimolar amounts of 3 and NaB(ArF)4 resulted in N2 evolution and clean formation of the diphenylcarbene complex salt [{(IPr)Ni}2(μ-Cl)(μ-CPh2)][B(ArF)4] (6) as forest-green crystals in 89% isolated yield (Scheme 3). Chloride abstraction with NaB(ArF)4 facilitated removal of neutral contaminants in a simple manner.
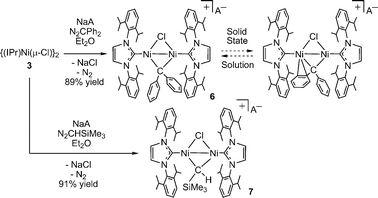 | ||
| Scheme 3 Synthesis of bridging, cationic carbene complexes 6 and 7 from diazoalkanes. | ||
The diphenylcarbene complex 6 is diamagnetic and exhibits a 1H-NMR spectrum consistent with C2v symmetry at both 195 K and room temperature. The bridging carbene-carbon of 6 resonates at δ 274 in the 13C-NMR spectrum (cf., δ 222 for the terminal carbene ligand in 1),2 and significantly downfield from the NHC-carbon attached to Ni (δ 177). The solid-state structure of 3, however, shows an unsymmetrical dimer in which the carbene functionality adopts η3-coordination involving the ipso- and one ortho-carbon of a phenyl ring along with the carbene carbon (see Fig. 1, Table 1). It is noteworthy that the Ni(2)–C(50) bond length is close to the value expected for a NiC double bond (cf., 1.836(2) Å in 1) while the Ni(1)–C(50) bond is significantly longer at 1.958(3) Å, and is closer to distances found for Ni–C single bonds in cationic nickel alkyls like [(dtbpe)Ni(CH2CMe2Ph)][PF6] with Ni–C = 1.954(3) Å.6
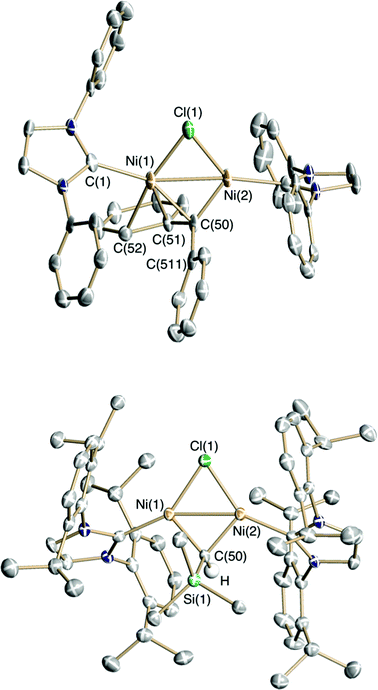 | ||
| Fig. 1 Solid-state structures of the complex cations of 6 (top) and 7 (bottom) drawn with 50% thermal ellipsoids. H-atoms (except on the carbene carbon), the B(ArF)4 anions, IPr iso-propyl groups (for 6), and co-crystallized solvent have been omitted for clarity. | ||
| 6 | 7 | ||||
|---|---|---|---|---|---|
| Atom 1 | Atom 2 | Dist./Å | Atom 1 | Atom 2 | Dist./Å |
| Ni(1) | Ni(2) | 2.4312(10) | Ni(1) | Ni(2) | 2.4385(9) |
| Ni(1) | C(50) | 1.958(3) | Ni(1) | C(50) | 1.849(3) |
| Ni(2) | C(50) | 1.831(5) | Ni(2) | C(50) | 1.858(3) |
| Ni(1) | C(1) | 1.975(5) | C(50) | Si(1) | 1.882(3) |
| Ni(1) | C(51) | 2.134(5) | |||
| Ni(1) | C(52) | 2.368(5) | |||
| C(50) | C(511) | 1.474(7) | |||
Reaction of N2CHSiMe3 with 3 in the presence of NaB(ArF)4 affords purple, crystalline [{(IPr)Ni}2(μ-Cl)(μ-CHSiMe3)][B(ArF)4] (7) in 91% isolated yield (Scheme 3). The highly deshielded carbene carbon of 7 resonates at δ 294 in the 13C-NMR spectrum, and the unique proton at δ 14.27 in the 1H-NMR spectrum. Carmona has reported several bridging Ni carbenes with “Ni2(μ-CHR)” cores that are formed via α–H elimination from a cyclometallated neophyl ligand in (PMe3)2Ni(CH2CMe2-o-C6H4), including examples with phosphine and cyclopentadienyl ancillary ligands.7 For comparison, Carmona's complexes exhibit bridging carbene-ligand resonances at δ 160–167 and δ 3.0–4.6 in their 13C- and 1H-NMR spectra, respectively.
A single-crystal X-ray study of 7 reveals a symmetrically bridging trimethylsilylcarbene ligand with Ni–C bond distances of 1.849(3) and 1.858(3) Å. The Ni–Ni distance of 7 is nearly identical to that in 6 despite the difference in carbene coordination mode (2.4312(10) vs. 2.4389(9) Å, respectively). Ni(1), Ni(2), Cl, and the carbene carbon are coplanar.
Polynuclear Ni complexes containing unsaturated carbyl fragments are relatively rare, primarily represented by bridging carbynes (LnNixCR),8 in addition to the aforementioned Carmona carbenes. The majority of these carbynes are trimeric species supported by CpNi subunits, exemplified by (CpNi)3(μ3-CCH3), a product of the reaction of excess LiCH3 with Cp2Ni.8e Heteronuclear trimetallic complexes have also been prepared by the reaction of Cp*W(CO)2(CR) with {(Cp)2Ni(μ-CO)}2, giving (CpNi)2Cp*W(μ3-CR)(CO)4.8c,d Kubiak has reported that N-protonation or alkylation of bridging isocyanide ligands affords [Ni2(μ-CNMeH)(CNMe)2(dppm)2]+ (dppm = bis(diphenylphosphino)methane) featuring bridging aminocarbyne moieties.9
Reactivity and group-transfer studies
In contrast to the tolerance of the imido dimers 4 and 5 toward excess mesitylazide (see above), both 3 and 6 readily convert excess N2CPh2 to a ∼ 1:1 mixture of Ph2CCPh2 and Ph2C–NN–CPh2 with elimination of dinitrogen (Scheme 4).10e Previous reports of both free and transition-metal-stabilized carbene and diazoalkane decomposition note the corresponding azine (in this case, Ph2CN–NCPh2) as the major product with the corresponding olefin (here, Ph2CCPh2) often constituting a minor component.10
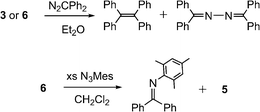 | ||
| Scheme 4 Reaction of N2CPh2 with 3 or 6 forms tetraphenylethylene and benzophenonediazine in a ∼ 1:1 product ratio. | ||
Reactions of 6 with mesityl azide, carbon monoxide, and t-butyl isocyanide were explored to elucidate its carbene group-transfer capabilities (see Schemes 4–6). It is noteworthy that Warren has reported related bridging dicopper carbenes displaying similar metric parameters which effect facile cyclopropenation and addition to suitable nucleophiles.11 As shown in Scheme 4, reaction of 6 with N3Mes results in its conversion to 5 with elimination of the ketimine Ph2CNMes, isolated in 80% yield. The reaction is slow, occurring over a period of three weeks at room temperature in the presence of excess N3Mes (CD2Cl2). Attempts to increase the reaction rate by heating to 60 °C resulted in rapid decomposition of 6. 1H-NMR studies of the room temperature reaction of 6 and N3Mes show the only nickel-containing product to be 5. We have reported on carbene–imide and carbene–oxo coupling with the nickel diphenylcarbene complex 1.4 Computational and kinetic studies indicate the formation of a 1,3-dipolar cycloaddition intermediate prior to N2 release. The slow reaction rate of 6 is possibly a consequence of steric inhibition of a related cycloaddition intermediate. Alternatively, the poor nucleophilicity of N3Mes may inhibit coordination to the sterically hindered Ni centers and slow formation of bridging μ-NMes or μ-N3Mes intermediates.
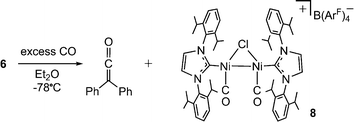 | ||
| Scheme 5 Combination of excess CO and 6 leads to formation of diphenylketene and the Ni(I)-Ni(I) dicarbonyl salt 8. | ||
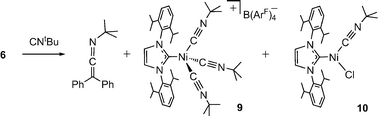 | ||
| Scheme 6 Addition of excess CNtBu to 6 forms Ph2CCNtBu with dissociation of the dimeric metal complex to Ni(I) monomers. | ||
Diphenylketene is readily formed by carbene-group transfer in the reaction of 6 with carbon monoxide. Exposure of 6 to an excess of CO results in clean conversion to free Ph2CCO and the diamagnetic, bridging Ni(I)-Ni(I) carbonyl dimer [{(IPr)Ni(CO)}2(μ-Cl)][B(ArF)4] (8, Scheme 5) as a purple salt. If the reaction is carried out in a 1:1 stoichiometry (6:CO), diphenylketene is formed in only ∼30% yield along with 8 and unreacted 6. Complex salt 8 is stable in the presence of 1 atm CO, and disproportionation to Ni(0) and Ni(II) is not observed. This is in contrast to the reaction of excess CO with 3 that results in disproportionation to the Ni(0) carbonyl complex (IPr)Ni(CO)3 and the Ni(II) dimer {(IPr)Ni(Cl)}2(μ-Cl)2.5 The solid-state structure of 8 (Fig. 2, Table 2) shows a symmetrical dimer with a carbonyl ligand binding in a κ1 fashion to each Ni(I) center. The Ni(1)–Ni(2) distance in 8 at 2.4967(5) Å is only slightly longer than the corresponding values found in 6 and 7. The carbonyl ligands bind to nickel in linear manners (Ni–C–O = 179.5(3) and 176.6(3)°) with C–O bond distances of 1.134(3) and 1.145(3) Å that indicate limited back-bonding. The IR data (νCO = 2063, 2023 cm−1) corroborate minimal activation of the carbonyl ligands in this cationic complex. For comparison, previously isolated examples of dimeric Ni(I) carbonyls exhibit νCO typically ranging from 1775–2020 cm−1.12
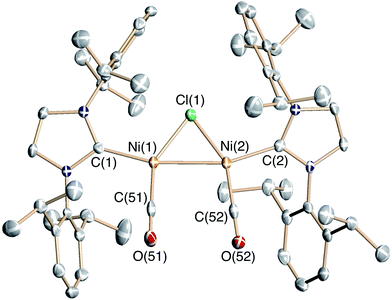 | ||
| Fig. 2 Solid-state structure of the complex cation of 8 drawn with 50% thermal ellipsoids. H-atoms, co-crystallized solvent, and the B(ArF)4 counterion have been omitted for clarity. | ||
| 8 | 9 | ||||
|---|---|---|---|---|---|
| Atom 1 | Atom 2 | Dist./Å | Atom 1 | Atom 2 | Dist/Å |
| Ni(1) | Ni(2) | 2.4967(5) | Ni(1) | C(41) | 1.916(3) |
| Ni(1) | C(1) | 1.943(3) | Ni(1) | C(42) | 1.906(3) |
| Ni(2) | C(2) | 1.958(3) | Ni(1) | C(43) | 1.919(3) |
| Ni(1) | C(51) | 1.762(3) | C(41) | N(41) | 1.152(4) |
| Ni(2) | C(52) | 1.754(3) | C(42) | N(42) | 1.164(4) |
| C(51) | O(51) | 1.134(3) | C(43) | N(43) | 1.160(4) |
| C(52) | O(52) | 1.145(3) | Ni(1) | C(1) | 1.988(3) |
Addition of an excess of tert-butyl isocyanide to a solution of 6 gives the carbene-coupling product Ph2CCNtBu in 76% isolated yield (Scheme 6). Although alkyl isocyanides often give metal products isostructural with the CO analogues, the fate of the Ni(I) fragment in this reaction differs from 8, undergoing dimer dissociation to give the mononuclear Ni(I) salt [(IPr)Ni(CNtBu)3][B(ArF)4] (9) and the known 3-coordinate Ni(I) complex (IPr)Ni(CNtBu)(Cl) (10).5 Complex 9 is a paramagnetic pale-yellow salt with μeff = 1.9 μB (Evans). The X-ray structure of 9 (Fig. 3, Table 2) shows a distorted tetrahedral Ni(I) center with three linear isocyanide ligands (Ni(1)-C(42)-N(42) = 169.4(3), Ni(1)-C(43)-N(43) = 176.1(3), Ni(1)-C(41)-N(41) = 169.2(3)°). When three equivalents of CNtBu are used, a mixture of 6, 9, and 10 is obtained (and not the dimeric pivaloisocyanide analogue of 8). The difference in the rate of reaction between CO, CNtBu, and N3Mes may be due to the ability of CO and CNtBu to readily coordinate to the Ni centers.
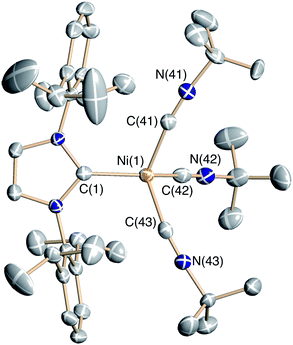 | ||
| Fig. 3 Solid-state structure of the complex cation of 9 drawn with 50% thermal ellipsoids. H-atoms, co-crystallized solvent, and the B(ArF)4 counterion have been omitted for clarity. | ||
Conclusions
Cationic, bridging dinickel carbene salts of the type [{(IPr)Ni}2(μ-Cl)(μ-CR1R2)][B(ArF)4] (6, R1 = R2 = Ph; 7, R1 = H, R2 = SiMe3) can be isolated in good yields from reactions of the Ni(I)-Ni(I) dimer {(IPr)Ni(μ-Cl)}2 (3) with diazoalkanes in the presence of NaB(ArF)4. Carbene transfer from 6 to the nitrene fragment “NMes” (from N3Mes), CO, and CNtBu was found to occur stoichiometrically to form Ph2CNMes, Ph2CCO, and Ph2CCNtBu, respectively. Exposure of 6 to excess carbon monoxide leads to the formation a Ni(I)-Ni(I) carbonyl dimer [{(IPr)Ni(CO)}2(μ-Cl)][B(ArF)4] (8) that is stable with respect to dimer dissociation. The related substrate CNtBu, however, effects dimer dissociation in its reaction with 6 with formation of the monomeric Ni(I) complexes [(IPr)Ni(CNtBu)3][B(ArF)4] (9) and (IPr)Ni(Cl)(CNtBu) (10).
Attempts to isolate the neutral carbene variant, {(IPr)Ni(Cl)}2(μ-CR1R2), were stymied by the competitive, facile decompostion of the diazoalkane substrate initiated by either 3 or the product carbene. This catalyticdiazoalkane decomposition (to the azine) prevents catalytic turnover for the stoichiometric carbene-transfer reactions we have explored since the substrate is readily consumed before productive carbene-transfer occurs.
Acknowledgements
This work was generously supported by the US National Science Foundation through Grant CHE-0957816 to G.L.H.Notes and references
- (a) E. O. Fisher and A. Maasböl, Angew. Chem., Int. Ed. Engl., 1964, 3, 580 CrossRef; (b) R. R. Schrock, Reactions of Coordinated Ligands; Braterman, P. S., Ed.; Plenum Press: New York, 1986 Search PubMed; (c) K. H. Dötz, Transition Metal Carbene Complexes; Verlag Chemie: Weinheim, Germany, 1983 Search PubMed; (d) R. R. Schrock, Angew. Chem., Int. Ed., 2006, 45, 3748 CrossRef CAS; (e) R. H. Grubbs, Angew. Chem., Int. Ed., 2006, 45, 3760 CrossRef CAS.
- D. J. Mindiola and G. L. Hillhouse, J. Am. Chem. Soc., 2002, 124, 9976 CrossRef CAS.
- (a) D. J. Mindiola and G. L. Hillhouse, Chem. Commun., 2002, 1840 RSC; (b) R. Waterman and G. L. Hillhouse, J. Am. Chem. Soc., 2003, 125, 13350 CrossRef CAS.
- N. D. Harrold, R. Waterman and G. L. Hillhouse, J. Am. Chem. Soc., 2009, 131, 12872 CrossRef CAS.
- C. A. Laskowski and G. L. Hillhouse, Organometallics, 2009, 28, 6114 CrossRef CAS.
- K. D. Kitiachvili, D. J. Mindiola and G. L. Hillhouse, J. Am. Chem. Soc., 2004, 126, 10554 CrossRef CAS.
- T. R. Balderrain, E. Gutierrez, A. Monge, M. C. Nicasio, M. Paneque, M. L. Poveda and E. Carmona, Organometallics, 1993, 12, 4431 CrossRef.
- Representative examples: (a) H. Beurich, R. Blumhofer and H. Vahrenkamp, Chem. Ber., 1982, 115, 2409 CAS; (b) R. Blumhofer, K. Fischer and H. Vahrenkamp, Chem. Ber., 1986, 119, 194 CAS; (c) G. P. Elliot, J. A. Howard, T. Mise, I. Moore, C. M. Nunn and F. G. A. Stone, J. Chem. Soc., Dalton Trans., 1986, 2091 RSC; (d) E. Delgado, J. Hein, J. C. Jeffery, A. L. Ratermann, F. G. A. Stone and L. J. Farrugia, J. Chem. Soc., Dalton Trans., 1987, 1191 RSC; (e) H. Lehmkuhl, C. Krüger, S. Pasynkiewicz and J. Poplawska, Organometallics, 1988, 7, 2038 CrossRef CAS.
- D. L. DeLaet, P. E. Fanwick and C. P. Kubiak, Organometallics, 1986, 5, 1807 CrossRef CAS.
- (a) W. E. Parham and W. R. Hasek, J. Am. Chem. Soc., 1954, 76, 935 CrossRef CAS; (b) C. Ruchardt and G. N. Schrauzer, Chem. Ber., 1960, 93, 1840 CrossRef CAS; (c) K. Ishiguro, Y. Sawaki and H. Iwamura, Chem. Lett., 1987, 9, 1853 CrossRef; (d) D. Griller, M. Majewski, W. G. McGimpsey, A. S. Nazran and J. C. Scaiano, J. Org. Chem., 1988, 53, 1550 CrossRef CAS; (e) P. Hofmann, I. V. Shishkov and F. Rominger, Inorg. Chem., 2008, 47, 11755 CrossRef CAS.
- Y. M. Badiei and T. M. Warren, J. Organomet. Chem., 2005, 690, 5989 CrossRef CAS.
- (a) R. A. Jones, A. L. Stuart, J. L. Atwood and W. E. Hunter, Organometallics, 1983, 2, 874 CrossRef CAS; (b) Z. Zhang, H. Wang, H. Wang, R. Wang, W. Zhao and L. Yang, J. Organomet. Chem., 1988, 347, 269 CrossRef CAS; (c) D. J. Elliot, D. G. Holah, A. N. Hughes, H. A. Mirza and E. Zawada, J. Chem. Soc., Chem. Commun., 1990, 32 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: All experimental protocols and complete crystallographic details for 6, 7, 8, and 9 (CIF) and data regarding the crystallographic refinements (PDF). CCDC reference numbers 791319–791322. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c0sc00464b |
| This journal is © The Royal Society of Chemistry 2011 |