Nitrogen-directed ketone hydroacylation: Enantioselective synthesis of benzoxazecinones†
Hasan A.
Khan
,
Kevin G. M.
Kou
and
Vy M.
Dong
*
Department of Chemistry, University of Toronto, 80 St. George Street, Toronto, Ontario M5S 3H6, Canada
First published on 30th November 2010
Abstract
We report a nitrogen-directed ketone hydroacylation to furnish 8-membered N-containing lactones (benzoxazecinones) in high yields and high enantiomeric excesses. In a model study, a comparison was made between nitrogen, oxygen and sulfur directing groups and it was found that nitrogen promoted faster hydroacylation. Moreover, the amine directing group completely suppressed competitive decarbonylation. This catalytic transformation generates a relatively unexplored class of nitrogen heterocycles, namely benzoxazepinones and benzoxazecinones.
Introduction
The vast majority of bioactive molecules and pharmaceutical agents consist of nitrogen-containing heterocycles.1 As a result, developing new ways to make nitrogen-containing heterocycles can positively impact medicinal chemistry and drug discovery. In general, transition metal-catalyzed routes to nitrogen-containing heterocycles can be difficult to access due to the propensity of amines to bind too strongly to metal centers. Catalytic and enantioselective strategies for making medium-sized rings are especially rare.2,3 By taking advantage of the Lewis basicity of amines, we herein demonstrate a ketone hydroacylation that provides seven and eight-membered nitrogen-containing heterocycles, respectively named benzoxazepinones and benzoxazecinones, in high efficiency and enantiomeric excess.Lewis-basic functional groups, specifically ethers and sulfides, are known to promote both intermolecular and intramolecular hydroacylations.4 These heteroatoms coordinate to the metal-center, and this coordination event aids C–H bond activation and suppresses decarbonylation. Our laboratory has reported asymmetric methods in which substrates, bearing oxygen or sulfur functionality, will undergo cyclization to produce medium-sized lactones and heterocyclic ketones.5 Although amine-promoted intramolecular hydroacylations have not been investigated, Bolm and co-workers have shown that 2-formylaniline derivatives can undergo intermolecular hydroacylation with norbornadiene to produce ketones in good yield, but with low enantioselectivity.6 To address this challenge, we propose an intramolecular amine-directed hydroacylation as shown in Scheme 1. The amine-assisted C–H bond activation would occur to generate a rhodabicycle that could undergo insertion and reductive elimination to produce medium-sized nitrogen-containing heterocycles.
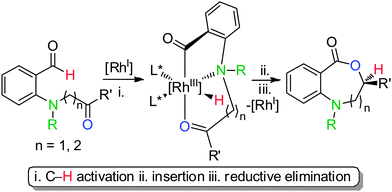 | ||
| Scheme 1 Proposed N-directed ketone hydroacylation towards chiral benzoxazepinones (n = 1) and benzoxazecinones (n = 2). | ||
Results and discussion
Initial studies
Our initial studies focused on the hydroacylation of substrates 1a–c, derived from commercially available methyl anthranilate, using our reported hydroacylation catalyst: [Rh((R)-DTBM-SEGPHOS)]BF4. With this model system, we were able to tune the basicity of the amine to achieve the desired transformation (Table 1). Strongly electron-withdrawing substituents (e.g., Ts or Ms) on the nitrogen atom led to no reactivity (entries 1 and 2). However, substrate 1c bearing a more Lewis basic N–Me group underwent lactonization to produce enantioenriched benzo[e][1,4]oxazepinone 2c in 86% yield and 72% ee.|
|
||||
|---|---|---|---|---|
| Entry | Substrate | t/min | Conv. 2 (%)b | ee (%)c |
| a Conditions: 2 mol% [Rh((R)-DTBM-SEGPHOS)]BF4, 0.2 mmol substrate (1), room temperature (rt), CH2Cl2. b Conversion determined by 1H-NMR. c Determined by chiral HPLC. d Starting material was recovered quantitatively. | ||||
| 1 | 1a X = N-Ts | — | 0d | — |
| 2 | 1b X = N-Ms | — | 0d | — |
| 3 | 1c1c X = N-Me | <5 | >99 | 72 |
| 4 | 1d X = O | 2880 | 36 | 99 |
| 5 | 1e X = S | 120 | >99 | 99 |
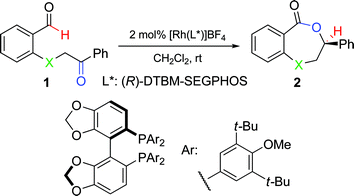
Next, we compared the reactivity of substrate 1c to the analogous O- or S-containing keto-aldehydes (Table 1, entries 4 and 5). By measuring conversion over time in the presence of 2 mol% [Rh((R)-DTBM-SEGPHOS]BF4, we observed marked differences in the relative rates for hydroacylation (Table 2). The O-bearing substrate achieved 36% conversion after 48 h, while the S-bearing keto-aldehyde underwent complete conversion to the desired lactone after 2 h. To our delight, N-bearing substrate 2c achieved >99% conversion in less than 5 min at room temperature. Hence, we were able to make use of alkylamines in intramolecular ketone hydroacylation viaRhI catalysis. Importantly, throughout these studies, no decarbonylation products were observed by1H-NMR in the N-directed hydroacylation reactions. Thus, the presence of a nitrogen atom not only promotes faster reactivity but also suppresses decarbonylation completely.
|
|
||||
|---|---|---|---|---|
| Entry | R | T/°C | Yield (%)a | ee (%)b |
| a Isolated yield. b Determined by chiral HPLC. | ||||
| 1 | C6H5 (1c) | 23 | 91 (2c) | 77 |
| 2 | C6H5 (1c) | −35 | 91 (2c) | 85 |
| 3 | n-Bu (1f) | −35 | 86 (2f) | 93 |
| 4 | CH3 (1g) | −35 | 85 (2g) | 75 |
| 5 | t-Bu (1h) | −35 | 90 (2h) | 50 |
Benzoxazepinone synthesis
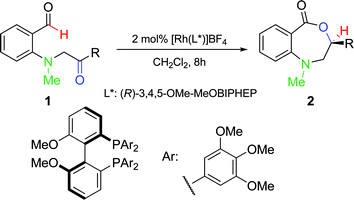
To improve the enantioselectivity, numerous chiral bidentate phosphine ligands were examined and (R)-3,4,5-OMe-MeOBIPHEP was found to offer a modest increase in ee. At −35 °C, the desired hydroacylation of keto-aldehyde 1c occurred in the presence of 2 mol% [Rh(R)-3,4,5-OMe-MeOBIPHEP)]BF4 in 85% ee and 91% yield (Table 2, entry 2). Under our optimized conditions, alkyl- and aryl-substituted keto-aldehydes undergo hydroacylation at low catalyst loading to produce the corresponding benzo[e][1,4]oxazepinones in high yields and moderate to high enantiomeric excesses (50–93% ee).Extending the size of the lactones
Having successfully synthesized chiral benzoazepinones, we next investigated the preparation of larger 8- and 9- membered lactones. Previous mechanistic studies provide support for the proposed [3.3.0]rhodabicycle intermediate prior to lactonization in the preparation of 7-membered lactones.4c We thus reasoned that substrates 3a and 5 could potentially proceed through a [3.4.0]- and [4.3.0]-rhodabicycle intermediate, respectively, in the catalytic cycle and give rise to chiral benzoxazecinones (Scheme 2). Indeed, aniline derivative 3a underwent intramolecular ketone hydroacylation in the presence of 2 mol% [Rh((R)-DTBM-SEGPHOS)]BF4 to give the corresponding benzoxazecinone 4a in 95% isolated yield and 96% ee, whereas benzylamine derivative 5 showed no reactivity even at higher catalyst loadings (up to 10 mol%) and elevated temperatures (up to 60 °C).7 Similarly, keto-aldehyde 6, which would theoretically proceed via a [4.4.0]-rhodabicycle to furnish a 9-membered ring benzoxazeninone, showed no reactivity. These results suggest that successful intramolecular ketone hydroacylation depends on both the basicity and the position of the heteroatom within the tether. Since it appeared that the 5-membered amino-acyl rhodacycle is critical to allow hydroacylation (3a → 4a), we envisioned substrates that would form [3.5.0]rhodacycle intermediates could provide chiral benzoxazeninone macrocycles following reductive elimination. However, subjecting substrate 7 to our ketone hydroacylation conditions also resulted in no observed reactivity.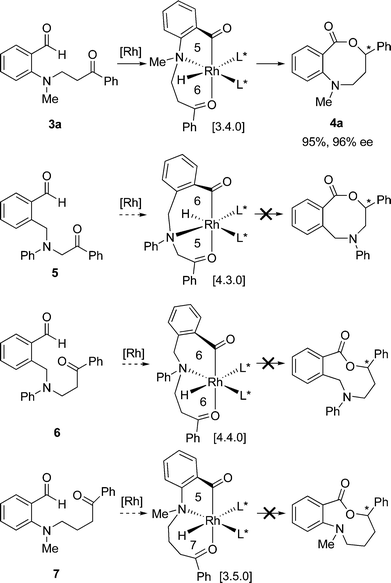 | ||
| Scheme 2 Attempted strategies in the preparation of larger size lactones. | ||
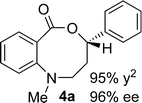
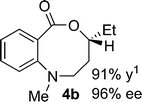
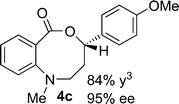
Synthesis and scope of benzoazecinones
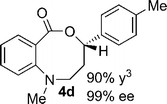
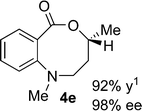
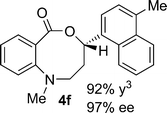
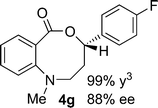
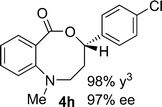
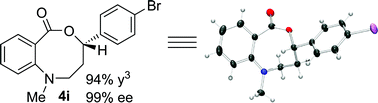
Structural analogues of benzo[e][1,4]oxazepinones are reported to have potent medicinal activity,8 and so it seemed likely that benzoxazecinones would also have some place in the literature of medicinal chemistry and natural products. We were surprised to learn, however, that these N-containing 8-membered lactones are unprecedented organic frameworks. We prepared a series of keto-aldehydes of the type 3 from commercially available N-methyl methylanthranilate. In the presence of [Rh((R)-DTBM-SEGPHOS)]BF4, these homologated keto-aldehydes underwent hydroacylation smoothly to provide the corresponding substituted benzo[c][1,5]oxazecinones 4 in high yields and enantioselectivities (Table 3, 84–99%, 88–99% ee). In contrast to the keto-aldehyes we presented in our earlier work with O-directing groups,3c both electron-donating and electron-withdrawing groups were accommodated. Notably, substrate 3c bearing an electron-rich arene substituent underwent hydroacylation to afford the corresponding 8-membered lactone 4c in 84% yield and 95% ee. As with the 7-membered lactones, no decarbonylation was observed. An X-ray crystal structure of lactone 4i allowed us to unambiguously determine the absolute configuration of the benzo[c][1,5]oxazecinones.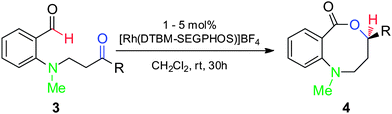
Conclusions
We have developed a catalytic lactonization to furnish chiral benzoxazepinones and benzoxazecinones in high yields and enantioselectivities. By using amines as a coordinating group, we observed more efficient hydroacylation as compared to the ether and sulfide analogues. This transformation is sensitive to both the coordinating ability of the amine and its position within the tether. Future work will focus on identifying more general catalysts and developing other amine-directed C–H bond activations for the preparation of nitrogen-containing heterocycles.Acknowledgements
We thank the University of Toronto, Canada Foundation for Innovation, Ontario Research Fund, and NSERC for funding. VMD is grateful for an Alfred P. Sloan Fellowship. HAK and KGMK acknowledge Edwin Walter & Margery Warren for OGSST fellowships. We appreciate the generous donation of ligands from Takasago ((R)-DTBM-SEGPHOS) and Solvias ((R)-3,4,5-OMe-MeOBIPHEP).Notes and References
- J. S. Carey, D. Laffan, C. Thomson and M. T. Williams, Org. Biomol. Chem., 2006, 4, 2337 RSC.
- T. E. Müller, K. C. Hultzsch, M. Yus, F. Foubelo and M. Tada, Chem. Rev., 2008, 108, 3795 CrossRef.
- For reviews of strategies for transition metal catalyzed syntheses of N-containing heterocycles, see: (a) G. Zeni and R. C. Larock, Chem. Rev., 2006, 106, 4644 CrossRef CAS; (b) P. Thansandote and M. Lautens, Chem.–Eur. J., 2009, 15, 5874 CrossRef CAS; (c) N. T. Patil and Y. Yamamoto, Chem. Rev., 2008, 108, 3395 CrossRef CAS; (d) G. Zeni and R. C. Larock, Chem. Rev., 2004, 104, 2285 CrossRef CAS; (e) Y. Troin and M.-E. Sinibaldi, in Asymmetric Synthesis of Nitrogen Heterocycles, ed. J. Royer, Wiley Verlag, Weinheim, 2009 Search PubMed; (f) B. M. Trost and M. J. Krische, Syn. Lett., 1998, 1.
- For examples of ethers and sulfides as chelating groups in intramolecular olefin hydroacylation, see: (a) M. C. Willis, Chem. Rev., 2010, 110, 725 CrossRef CAS; (b) K. Tanaka and G. C. Fu, J. Am. Chem. Soc., 2003, 125, 8078 CrossRef CAS; (c) K. Takeishi, K. Sugishima, K. Sasaki and K. Tanaka, Chem.–Eur. J., 2004, 10, 5681 CrossRef CAS; (d) H. D. Bendorf, C. M. Colella, E. C. Dixon, M. Marchetti, A. N. Matukonis, J. D. Musselman and T. A. Tiley, Tetrahedron Lett., 2002, 43, 7031 CrossRef CAS. For examples of ethers and sulfides in intermolecular olefin hydroacylation, see: (e) J. D. Osbourne, H. E. Randell-Sly, G. S. Currie, A. R. Cowley and M. C. Willis, J. Am. Chem. Soc., 2008, 130, 17232 CrossRef; (f) Y. Shibata and K. Tanaka, J. Am. Chem. Soc., 2009, 131, 12552 CrossRef CAS; (g) D. T. H. Phan, K. G. M. Kou and V. M. Dong, J. Am. Chem. Soc., 2010 DOI:10.1021/ja107738a; (h) M. M. Coulter, K. G. M. Kou, B. Galligan and V. M. Dong, J. Am. Chem. Soc., 2010 DOI:10.1021/ja107198e.
- (a) Z. Shen, H. A. Khan and V. M. Dong, J. Am. Chem. Soc., 2008, 130, 2916 CrossRef CAS; (b) D. H. T. Phan, B. Kim and V. M. Dong, J. Am. Chem. Soc., 2009, 131, 15608 CrossRef CAS; (c) Z. Shen, P. K. Dornan, H. A. Khan, T. K. Woo and V. M. Dong, J. Am. Chem. Soc., 2009, 131, 1107; (d) M. M. Coulter, P. K. Dornan and V. M. Dong, J. Am. Chem. Soc., 2009, 131, 6932 CrossRef CAS.
- For an example of anilines as directing groups in intermolecular olefin hydroacylation, see: (a) R. T. Stemmler and C. Bolm, Adv. Synth. Catal., 2007, 349, 1185 CrossRef CAS. For examples of pyridyl-N as directing group in intermolecular olefin hydroacylation, see: (b) J. W. Suggs, J. Am. Chem. Soc., 1979, 101, 489 CrossRef CAS; (c) M. C. Willis and S. Sapmaz, Chem. Commun., 2001, 2558 RSC; (d) Y. J. Park, J.-W. Park and C.-H. Jun, Acc. Chem. Res., 2008, 41, 222 CrossRef CAS.
- Trace amounts of the undesired decarbonylation product were observed by 1H NMR spectroscopy at 120 °C.
- (a) A. J. Cocuzza, D. R. Chidester, B. C. Cordova, R. M. Klabe, S. Jeffrey, S. Diamond, C. A. Weigelt, S. S. Ko, L. T. Bacheler, S. K. Erickson-Viitanen and J. D. Rodgers, Bioorg. Med. Chem. Lett., 2001, 11, 1389 CrossRef CAS; (b) L. Banfi, A. B. G. Guanti, P. Lecinska and R. Riva, Org. Biomol. Chem., 2006, 4, 4236 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: General procedures, spectral data and X-ray crystal structures. CCDC reference number 791672. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c0sc00469c |
| This journal is © The Royal Society of Chemistry 2011 |