Bending contorted hexabenzocoronene into a bowl†
Adam C.
Whalley
a,
Kyle N.
Plunkett
b,
Alon A.
Gorodetsky
a,
Christine L.
Schenck
a,
Chien-Yang
Chiu
a,
Michael L.
Steigerwald
*a and
Colin
Nuckolls
*a
aDepartment of Chemistry, Columbia University, New York, NY, USA. E-mail: cn37@columbia.edu; mls2064@columbia.edu; Fax: +212 932 1289; Tel: +212 854 6289
bDepartment of Chemistry, Southern Illinois University, Carbondale, IL, USA
First published on 11th October 2010
Abstract
This article describes the synthesis of a new type of bowl-shaped polycyclic aromatic hydrocarbon. These bowls are formed by joining the proximal carbons of contorted hexabenzocoronenes. These methods begin to tap a wealth of structural diversity available from these core structures. The bowl-shaped hydrocarbons more easily accept electrons than their contorted hexabenzocoronene precursors and associate strongly with C70.
This article describes the preparation and photophysical properties of a new type of bowl-shaped polycyclic aromatic hydrocarbon (PAH) formed by joining the proximal carbons of contorted hexabenzocoronenes. Contorted hexabenzocoronenes1 are a class of compounds that have six 4-carbon annulations circumferential to coronene (1). This structure is highly non-planar as a result of the steric congestion around its periphery. Previously, we found these molecules dehydrogenate upon heating on a ruthenium surface to form highly curved structures.2 The main dehydrogenated product was assigned a structure in which six 5-membered rings had formed between its proximal carbons. These compounds are interesting because they represent a new class of bowl-shaped PAHs.3–7 Unfortunately, the resulting bowl-shaped molecules are so tightly bound to the metal surface that they could not be freed from the surface for further study. Here we develop the solution-based chemistry to synthesize and study two members of this class of PAHs with two and four of their five-membered rings formed (2 and 3, respectively). We find that the resulting compounds are highly non-planar, are bowl-shaped, and complex strongly with C70.
Fig. 1 shows the structural diversity resulting from the closure of the five-membered rings around the coronene core: twelve structural siblings that differ in the quantity and position of the five-membered rings. In addition, several of the compounds can exist in multiple conformations. The molecular shape of each analog varies significantly with subsequent five-membered ring incorporation. We focused this study on 2 and 3 because they were synthetically accessible and DFT calculations predicted the emblematic bowl architecture. Furthermore, our earlier computational studies8 showed that the compounds with four five-membered rings closed are the most energetically disfavored of the compounds in Fig. 1 due to their high degree of curvature.
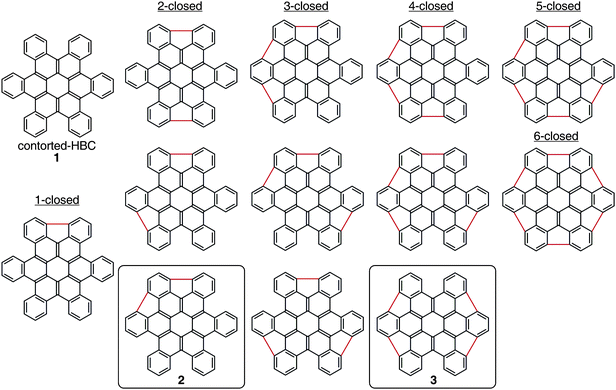 | ||
| Fig. 1 Illustration of the structural diversity available via closure of the 5-membered rings around the exterior of hexabenzocoronene (1). Derivatives of the structures highlighted with boxes (2, 3) were synthesized in this study. | ||
The DFT-optimized structures of 2 and 3 (B3LYP functional with the 631G** basis set) are shown in Fig. 2 and 3, respectively. Similar to previously crystallized contorted HBCs,1 the three “free” annulated benzo-substituents in 2 (rings b, c and b′ in Fig. 2A) are contorted and follow a down-up-down arrangement. The conformational isomer where the arrangement of b, c, and b′ is up-down-up is ∼7.5 kcal mol−1 higher in energy than the structure in Fig. 2. The bowl of 2 is composed of three annulated benzene rings connected through two five-membered rings and is concave above the two adjacent benzene rings. The splay angle (defined in Fig. 2A) between the a/b rings (38°) and the b/c rings (39°) provides a measure for the degree of strain inherent to these subunits. For comparison, the exterior ring splay for 1 is 42° while that for 4-helicene is 31°.9 We conclude that the newly formed five-membered rings pull the benzo-substituents back to alleviate some of the steric congestion around the periphery. These rings are further pulled back in 3 where the splay angle between the two rings that are not joined by five-membered rings is a mere ∼3°. The locus of the majority of strain in 3 is in its bowl-shaped portions and not in the 4-helicene subunits.
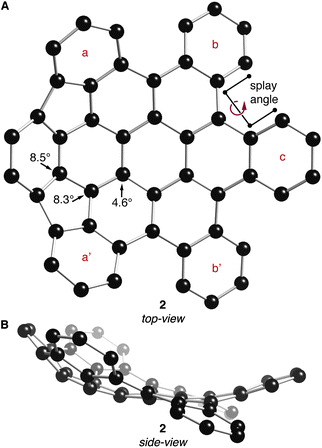 | ||
| Fig. 2 (A) Top- and (B) side-view of the DFT calculated structure of 2. POAV angles at their corresponding carbon atoms. Carbons are shown with black spheres. Hydrogen atoms have been removed for clarity. | ||
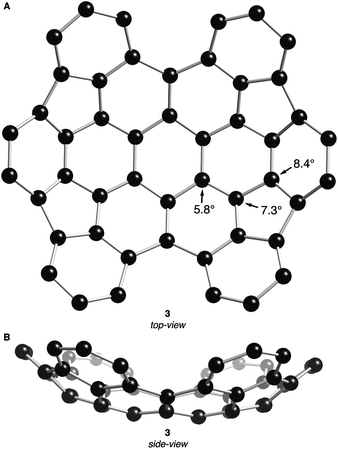 | ||
| Fig. 3 (A) Top- and (B) side-view DFT calculated structure of 3. POAV angles at their corresponding carbon atoms. Carbons are shown with black spheres. Hydrogen atoms have been removed for clarity. | ||
The bowl in 2 has a high degree of curvature and the π-orbital axis vector (POAV) pyramidalization angles,10,11 defined as θσπ – 90°, were calculated to be 8.5°, 8.3°, and 4.6° for the most distorted carbon atoms in the central area of the bowl in 2 (shown in Fig. 2A). These values are slightly greater than those of corannulene (8.2°)12 but less than C60 (11.6°).11 Compound 3 contains two cupped regions that slightly increased the strain on the most interior carbons with the POAV angles calculated to be 8.4°, 7.3°, and 5.8° (shown in Fig. 3A).
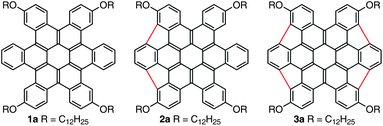
Our syntheses of 2 and 3 relied on n-dodecyloxy hydrocarbon chains (2a and 3a) to ensure solubility throughout. These compounds were derived from HBC precursors that would allow us to employ the solution-based, palladium-catalyzed chemistry, which was pioneered by Scott4 and Shevlin,3,13 to form the strained, five-membered rings. These heavily substituted HBC derivatives (5a,b) were synthesized from the corresponding halogenated pentacenequinone cores (4a,b) as shown in Scheme 1.14,15 For the final ring closure, converting 5a to 2a, we exposed the compound to microwave-assisted Heck conditions using [Pd(PCy3)2Cl2] as the catalyst and DBU as the base. The double ring-closure proceeded smoothly and 2a was isolated as a red solid in 82% yield. In a similar reaction with 5b, 3a formed in good yield (as indicated by 1H-NMR), but it was difficult to separate 3a from the derivative in which only three of the 5-membered rings formed, the fourth position being simply dehalogenated.16 Successive chromatographic separations gave pure 3a as a red solid in 16% yield.
![Synthesis of 2a and 3a. Key: (a) CBr4, PPh3, toluene, 80 °C, 16 h. (b) [Pd(PPh3)4], 2-(4-(dodecyloxy)phenyl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane, THF, 2 M K2CO3 (aq), 100 °C, 16 h. (c) hν, I2, benzene, propylene oxide, 3 h, rt. (d) FeCl3, CH3NO2, CH2Cl2, 3 h, rt. (e) [Pd(PCy3)2Cl2], DBU, DMA, μwave, 150 °C, 3 h.](/image/article/2011/SC/c0sc00470g/c0sc00470g-s1.gif) | ||
| Scheme 1 Synthesis of 2a and 3a. Key: (a) CBr4, PPh3, toluene, 80 °C, 16 h. (b) [Pd(PPh3)4], 2-(4-(dodecyloxy)phenyl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane, THF, 2 M K2CO3 (aq), 100 °C, 16 h. (c) hν, I2, benzene, propylene oxide, 3 h, rt. (d) FeCl3, CH3NO2, CH2Cl2, 3 h, rt. (e) [Pd(PCy3)2Cl2], DBU, DMA, μwave, 150 °C, 3 h. | ||
The thermal inversion barriers for the bowls are quite high. By observing the coalescence of diastereotopic methylene hydrogens with variable temperature NMR, we determined an inversion barrier of 18.5 kcal mol−1 in 2a (complete coalescence occurs at 95 °C). As a point of comparison the tetra-dodecyloxy HBC (1a) inversion barrier is 16.7 kcal mol−1 (65 °C).173a also showed diastereotopic methylene hydrogens; however, we observed no coalescence even upon heating to 180 °C. This suggests that 3a is conformationally locked and unable to invert on the NMR timescale at 180 °C.18
HBCs are generally yellow whereas the bowls, 2a and 3a, are red. This shift in color suggests that the induced curvature substantially changes the electronic structure. The UV-vis spectra of compounds 1a–3a can be seen in Fig. 4. Compound 1a has a typical absorbance with a λmax at 380 nm and only weak absorbance above 425 nm.1,19 Compound 2a shows a very slight blue-shift of the λmax to 377 nm but the longer-wavelength transition gains significant intensity. Compound 3a shows an overall red-shift of both the λmax to 392 nm as well as a broad absorbance above 425 nm. The sequential closures of the five-membered rings do not have a monotonic effect on the position of the lowest-energy optical absorptions of 1a–3a as 2a's absorption extends to the longest wavelength of the three.
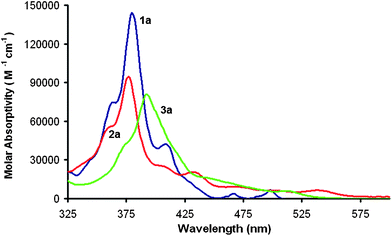 | ||
| Fig. 4 UV-vis spectra of 1a (blue), 2a (red), and 3a (green) in CH2Cl2. Concentration = 3.00 × 10−6 M, l = 1 cm. | ||
To understand this trend in the UV-vis absorbance, we undertook electronic structure calculations. Time-dependent DFT calculations (modeling the tetramethoxy analogs) showed that in all three molecules the lowest-energy, spin-allowed excitations are primarily from the HOMO to the LUMO. In each case, the HOMO is one of the radialene π set, but while the LUMO of 1 is one of the radialene π* orbitals, the LUMO in 2 and 3 are closely associated with the new five-membered rings. This suggests the development of cyclopentadienide character in the new rings, a suggestion that is substantiated by the decrease in LUMO energies (−1.6 eV, −2.0 eV, and −2.1 eV in 1, 2, and 3, respectively). This trend alone would predict that 3 would absorb to the red of 2, however the trend in HOMO energies (−4.8 eV, −4.8 eV, and −5.0 eV) belies this. The HOMO energy in 3 is lowered because the two radialene R2C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CR2 arrays that host the HOMO are closer to planar than are the corresponding sites in 1 and 2 (See Supporting Information). Thus while closing the five-membered rings does monotonically lower the energy of the LUMO, the effect on the HOMO is not so uniform.
CR2 arrays that host the HOMO are closer to planar than are the corresponding sites in 1 and 2 (See Supporting Information). Thus while closing the five-membered rings does monotonically lower the energy of the LUMO, the effect on the HOMO is not so uniform.
Cyclic voltammetry (see Table 1) supports the conclusions for the calculated energies of 1a–3a. The oxidation potentials of 1a and 2a are roughly the same, but that of 3a is significantly higher. This is the same trend that we observe with the HOMO energies from DFT. The reduction potentials similarly map the LUMO landscape: the highest energy reduction is roughly equivalent throughout the three molecules and represents reduction of the core. Additionally, for each aromatic five-membered ring there is an additional low-energy reduction: 2a shows two reductions (core plus one cyclopentadienide), and 3a shows three reductions (core plus two cyclopentadienides). These results further exemplify the ability of bowl-like structures to stabilize negative charges.20 This feature hints that these materials may be useful as n-type organic semiconductors in thin film devices.
The fluorescence emission spectra of compounds 1a–3a show a dramatic reduction in light emission, as well as a general red shift, when the number of five-membered rings increases (Fig. 5A). While 1a is highly fluorescent with a quantum yield of 0.14 (CH2Cl2), the quantum yields of 2a and 3a were determined to be 0.034 and 0.0085 (CH2Cl2), respectively. The apparent fluorescence dampening is reminiscent of fullerenes, which have quantum yields on the order of 1 × 10−4, as well as other small molecules containing five-membered rings that display no detectable fluorescence.20
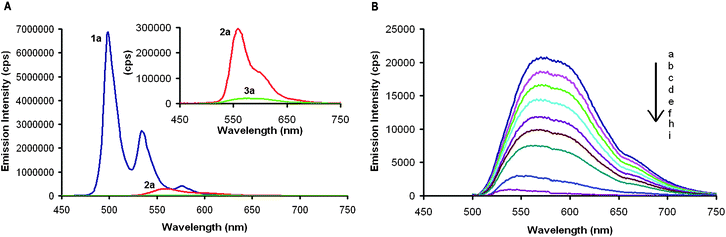 | ||
| Fig. 5 (A) Fluorescence emission spectra of 1a (blue), 2a (red), and 3a (green) at 1.00 × 10−6 M in CH2Cl2. Inset shows the zoomed in region of 2a and 3a. (B) Fluorescence emission spectra of 3a (1.00 × 10−6 M in CH2Cl2) excited at 392 nm with: (a) 0, (b) 0.05, (c) 0.10, (d) 0.15, (e) 0.2, (f) 0.25, (g) 0.3, (h) 0.4, and (i) 1.0 molar equivalents of C70 added. | ||
Recently, we showed that HBC co-crystallized with C60 in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 or 1:1 stoichiometry in the solid state.21 The favorable interactions between the π-face of the contorted HBC and the fullerene drive this co-crystallization. 2a and 3a have a more defined bowl-like structure that should allow them to interact with fullerenes more intimately. We initially chose C70 as a guest molecule as it has an oval shape that is more shape complementary to 3a. Our test for binding was to monitor the fluorescence quenching when equimolar amounts of C70 were added to 1a–3a. No measurable decrease in emission intensity was found for C70 and 1a at equimolar concentrations in CH2Cl2 (Supporting Information), suggesting that the association between 1 and C70 is minimal in solution. 2a emission is quenched by about 50% when it is mixed with equimolar amounts of C70 (Supporting Information). 3a shows completely quenched fluorescence when ∼0.5 equivalents of C70 is added (Fig. 5B). This demonstrates that the association constants between 2a and C70 as well as 3a and C70 are very high.22–25 Using a simple Stern–Volmer analysis, an association constant was calculated to be 4.7 × 105 M−1 for 2a and C70 and 3.2 × 106 M−1 for 3a and C70.26
:2 or 1:1 stoichiometry in the solid state.21 The favorable interactions between the π-face of the contorted HBC and the fullerene drive this co-crystallization. 2a and 3a have a more defined bowl-like structure that should allow them to interact with fullerenes more intimately. We initially chose C70 as a guest molecule as it has an oval shape that is more shape complementary to 3a. Our test for binding was to monitor the fluorescence quenching when equimolar amounts of C70 were added to 1a–3a. No measurable decrease in emission intensity was found for C70 and 1a at equimolar concentrations in CH2Cl2 (Supporting Information), suggesting that the association between 1 and C70 is minimal in solution. 2a emission is quenched by about 50% when it is mixed with equimolar amounts of C70 (Supporting Information). 3a shows completely quenched fluorescence when ∼0.5 equivalents of C70 is added (Fig. 5B). This demonstrates that the association constants between 2a and C70 as well as 3a and C70 are very high.22–25 Using a simple Stern–Volmer analysis, an association constant was calculated to be 4.7 × 105 M−1 for 2a and C70 and 3.2 × 106 M−1 for 3a and C70.26
Conclusions
In conclusion, we have synthesized a new class of bowl-shaped PAHs based on a coronene core. The shallow bowl shape of 3a shows an enhanced ability to stabilize negative charge and to bind C70 very strongly. The unique optical, electronic, and structural properties of these new bowl-shaped PAHs provide opportunities to create new n-type organic materials, phase-compatibilizers between p-type semiconductors and fullerenes in photovoltaic devices, and novel host–guest complexes.Acknowledgements
This research was funded by the Chemical Sciences, Geosciences and Biosciences Division, Office of Basic Energy Sciences, US D.O.E. (#DE-FG02-01ER15264), by the Center for Re-Defining Photovoltaic Efficiency Through Molecule Scale Control, an Energy Frontier Research Center funded by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences under Award Number DESC0001085, and by the FCRP FENA program. C.L.S. wishes to acknowledge support by the NSF GRFP; A.A.G. was supported by the National Science Foundation under Award Number 0936923; and A.C.W. acknowledges the support of the Guthikonda Fellowship in Organic Chemistry.Notes and references
- S. Xiao, M. Myers, Q. Miao, S. Sanaur, K. Pang, M. L. Steigerwald and C. Nuckolls, Angew. Chem., Int. Ed., 2005, 44, 7390–7394 CrossRef CAS.
- K. T. Rim, M. Siaj, M. Myers, L. Liu, C. Su, M. L. Steigerwald, S. Hybertsen, P. H. McBreen, G. W. Flynn and C. Nuckolls, Angew. Chem., Int. Ed., 2007, 46, 7891–7895 CrossRef CAS.
- L. Wang and P. B. Shevlin, Org. Lett., 2000, 2, 3703–3705 CrossRef CAS.
- H. A. Reisch, M. S. Bratcher and L. T. Scott, Org. Lett., 2000, 2, 1427–1430 CrossRef.
- T. Amaya, T. Nakata and T. Hirao, J. Am. Chem. Soc., 2009, 131, 10810–10811 CrossRef CAS.
- B. D. Steinberg, E. A. Jackson, A. S. Filatov, A. Wakamiya, M. A. Petrukhina and L. T. Scott, J. Am. Chem. Soc., 2009, 131, 10537–10545 CrossRef CAS.
- H. Sakurai, T. Daiko, H. Sakane, T. Amaya and T. Hirao, J. Am. Chem. Soc., 2005, 127, 11580–11581 CrossRef CAS.
- Unpublished data.
- F. H. Herbstein and G. M. J. Schmidt, J. Chem. Soc., 1954, 3302–3313 RSC.
- R. C. Haddon and L. T. Scott, Pure Appl. Chem., 1986, 58, 137–142 CAS.
- R. C. Haddon, J. Am. Chem. Soc., 1990, 112, 3385–3389 CrossRef CAS.
- D. Bakowies and W. Thiel, J. Am. Chem. Soc., 1991, 113, 3704–3714 CrossRef CAS.
- L. Wang and P. B. Shevlin, Tetrahedron Lett., 2000, 41, 285–288 CrossRef CAS.
- C. R. Swartz, S. R. Parkin, J. E. Bullock and J. E. Anthony, Org. Lett., 2005, 7, 3163–3166 CrossRef CAS.
- K. N. Plunkett, K. Godula, C. Nuckolls, N. Tremblay, A. C. Whalley and S. Xiao, Org. Lett., 2009, 11, 2225–2228 CrossRef CAS.
- The 3-closed HBC contaminant was evident viaNMR, but we were unable to obtain an analytically pure sample.
- T. J. Seiders, K. K. Baldridge, G. H. Grube and J. S. Siegel, J. Am. Chem. Soc., 2001, 123, 517–525 CrossRef.
- Analogs of 1a, 2a, and 3a were synthesized with two alkyl chains in order to simplify analysis of the variable temperature 1H NMR data (see supplemental information).
- Y. S. Cohen, S. Xiao, M. L. Steigerwald, C. Nuckolls and C. R. Kagan, Nano Lett., 2006, 6, 2838–2841 CrossRef CAS.
- H. Dang, M. Levitus and M. A. Garcia-Caribay, J. Am. Chem. Soc., 2002, 124, 136–143 CrossRef CAS.
- N. J. Tremblay, A. A. Gorodetsky, M. Cox, T. Schiros, B. Kim, R. Steiner, Z. Bullard, A. Sattler, W.-Y. So, Y. Itoh, M. F. Toney, H. Ogasawara, A. P. Ramirez, I. Kymissis, M. L. Steigerwald and C. Nuckolls, ChemPhysChem, 2010, 11, 799–803 CrossRef CAS.
- A. Sygula, F. R. Fronczek, R. Sygula, P. W. Rabideau and M. M. Olmstead, J. Am. Chem. Soc., 2007, 129, 3842–3843 CrossRef CAS.
- C. Mück-Lichtenfeld, S. Grimme, L. Kobryn and A. Sygula, Phys. Chem. Chem. Phys., 2010, 12, 7091–7097 RSC.
- P. E. Georghiou, L. N. Dawe, H.-A. Tran, J. Strübe, B. Neumann, H.-G. Stammler and D. Kuck, J. Org. Chem., 2008, 73, 9040–9047 CrossRef CAS.
- A. Molina-Ontoria, G. Fernádez, M. Wielopolski, C. Atienza, L. Sánchez, A. Gouloumis, T. Clark, N. Martín and D. M. Guldi, J. Am. Chem. Soc., 2009, 131, 12218–12229 CrossRef CAS.
- V. O. Stern and M. Volmer, Physik. Zeitschr., 1919, 20, 183–188 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental and computational details. See DOI: 10.1039/c0sc00470g |
| This journal is © The Royal Society of Chemistry 2011 |