Synthesis of boron doped polymeric carbon nitride solids and their use as metal-free catalysts for aliphatic C–H bond oxidation†
Yong
Wang
*a,
Haoran
Li
b,
Jia
Yao
b,
Xinchen
Wang
a and
Markus
Antonietti
a
aDepartment of Colloid Chemistry, Max-Planck Institute of Colloids and Interfaces, Research Campus Golm, D-14424, Potsdam, Germany. E-mail: Yong.Wang@mpikg.mpg.de
bDepartment of Chemistry, Zhejiang University, 310027, Hangzhou, P.R. China. E-mail: chemwy@zju.edu.cn
First published on 3rd December 2010
Abstract
Oxidation of hydrocarbons with clean oxidants is a crucial process for the development of products ranging from commodity chemicals to speciality pharmaceuticals. While selective oxidation is commonplace in nature and mainly occurs through a broad variety of cytochrome P450 enzymes, the synthetic catalysts that have been identified so far are relatively inactive with aliphatic C–H bonds. We present here a promising strategy for modifying polymeric graphitic carbon nitride by replacing the carbon atoms in the “melon” network using boron. 11B solid-state MAS NMR and XPS spectral studies confirmed the incorporation of the boron atoms in the polymeric g-C3N4 as well as the degree of doping, while X-ray diffraction and IR indicated that the characteristic structural properties of polymeric g-C3N4, such as the original graphitic structure, and the C–N heterocycle units were essentially retained. The boron-doped polymeric carbon nitride shows promising catalytic activity and high selectivity (≥87%) in the oxidation of benzylic aromatics. The use of this metal-free, organic, semiconducting polymer as an oxidation catalyst and H2O2 or O2 as oxidants makes the reactions interesting from both an economic and environmental point of view. It is also hoped that our findings will stimulate further investigation and will open new possibilities to form hybrid carbon nitride materials with new and probably exciting applications.
Introduction
Oxidation, especially oxidation of aliphatic (sp3) C–H bonds is a core technology for converting petrochemical based materials to commodity chemicals in the chemical industry.1–5 However, selective catalytic oxidation of sp3 hybridized carbons, within hydrocarbons, to alcohols, aldehydes, or ketones remains a challenging topic in contemporary chemical research.6–9 The increased reactivity of the products relative to the starting material makes selective oxidation very difficult. The catalysts available today for heterogeneous oxidation are generally far from ideal. Industrially performed catalytic oxidation reactions often suffer from drawbacks such as poor selectivity due to over-oxidation, lack of catalyst recycling, and negative environmental impact due to the use of toxic metal catalysts, etc.10–13 For example, in industry oxidation of toluene is achieved by using heavy metal based catalysts at high temperature (>200 °C). Generally, the conversion has to be kept at less than 4% to attain 70% selectivity of benzaldehyde and to avoid the formation of the carboxylic acid.14,15 The oxidation catalysts of the future have to be highly selective, environmentally benign and highly stable. Investigations into exquisitely selective biological catalysts, such as the oxidase and oxygenase enzymes, and some biomimetic oxidative catalysts are guiding us towards the ideal oxidation catalyst.16–21 Due to the efficiency of enzymes, the development of molecular compounds that can mimic the reactivity of enzymes have been extensively studied in recent years.22–29 However, most of the biomimetic catalysts that have been identified so far are relatively inactive toward aliphatic C–H bonds, and catalyst preparation often remains critical. Moreover, reducing agents are usually needed to compensate the excessive oxidation.30,31 Therefore, alternative metal-free heterogeneous catalysts for the selective oxidation of aliphatic C–H bond are highly desirable.In our search for biomimetic systems, we focus on heterogeneous organocatalysis, essentially adjusting the HOMO and LUMO positions and the coupled redox properties along with the natural models. Recently, a nitrogen-rich graphitic carbon nitride (g-C3N4) material has attracted much attention.32–37 This material has a heterocyclic macrocycle structure with a N–C–N– bonding pattern resembling a porphyrin (Scheme S1 ESI†). It was found that this polymeric material exhibits high chemical and thermal stability and an appealing electronic structure, being a wide-band gap, indirect semiconductor.38 This allows its direct use in heterogeneous catalysis.8,39–41 In addition, the catalytic, electronic and optical properties of the structure are by principle adjustable, using the rich chemistry of carbon and nitrogen.42–45 Indeed, similar “doping” protocols have been widely used to control the performance of more traditional materials. For example, various lightweight materials with diverse nanomorphologies that contain heteroatoms such as nitrogen, boron, or fluorine, have been actively pursued because of their unusual properties, say as semiconductors or in catalytic applications.46,47 Among the different non-metal dopants, boron seems to be especially promising in the materials field.48,49 A promising strategy for modifying g-C3N4 exists by replacing the C atoms in the network by boron to form a π-bonded, planar layered configuration like melon (Scheme 1). The active site, the heterocyclic macrocycle structure, is still there and the boron functional groups on the surface might act as strong Lewis acid sites, thus complementing the basic nitrogen sites in a bifunctional catalysis.
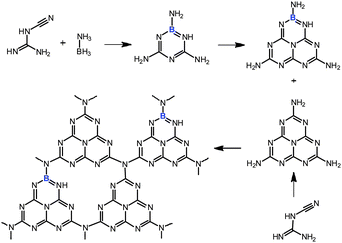 | ||
| Scheme 1 Proposed copolymerization processes of dicyandiamide with BH3NH3 at high temperature. | ||
We herein demonstrated the synthesis of boron-doped polymeric carbon nitride (CNB) solids using BH3NH3 as a molecular doping source which avoids the use of oxygen-containing precursors or the generation of HF, as with BF4−.8,43 We also demonstrate the catalytic activity of these CNB materials in the heterogeneous, metal-free oxidation of aliphatic C–H bonds. The CNB materials show superior performance (as compared to the previous homogeneous biomimetic oxidation catalysts) in the oxidation of toluene and other substituted aromatics with good conversion and an excellent aldehyde or ketone selectivity.
Experimental
Materials and methods
Unless otherwise stated, all solvents and chemicals used were of commercially available analytical grade. WAXS spectra were recorded on a Bruker D8 Advance diffractometer. UV-Vis spectra were recorded on a Varian Cary 4E UV-Vis system equipped with a Labsphere diffuse reflectance accessory. FTIR spectra were recorded on a BioRad FTS 6000 spectrometer, equipped with an attenuated total reflection (ATR) setup. 11B solid-state MAS NMR was recorded on a Bruker Avance III 400M NMR. X-ray photoelectron spectroscopy (XPS) data were obtained on Thermo ESCALAB250 instrument with a monochromatized Al-Kα line source (200 W). All binding energies were referenced to the C1s peak at 288.2 eV39 corresponding to a C–N–C coordination in CNB.Synthetic procedures for CNBx materials
The synthesis of CNB samples has been carried out employing a directly incorporating of different amount of BH3NH3 (x = 0.01, 0.025, 0.05, 0.1, 0.15, 0.20, 0.25 g, where x is the exact weight of BH3NH3 used per 3 g dicyandiamide) into the classical carbon nitride condensation (the resulting samples are denoted CNBx). In a typical synthetic procedure, BH3NH3 (0.15 g) was dissolved in 10 ml water and stirred for 5 min. Then 3 g dicyandiamide was added into the aqueous solution and the mixture was heated in an oil bath at 80 °C until the water was removed and a white solid formed. The white solid was then transferred into a crucible and heated for 4 h to reach a temperature of 600 °C and tempered at this temperature for 4 h under high purity nitrogen with a flow rate of 500 ml min−1. The sample was then allowed to cool in the oven to room temperature.Typical procedure for the catalytic oxidation of aromatics
The oxidation of toluene or ethylbenzene over a CNBx sample using H2O2 as the oxidant was carried out in a teflon-lined stainless steel batch reactor (40 ml total volume), using acetonitrile as the reaction solvent. Toluene or ethylbenzene (0.8 ml), catalyst (50 mg), 30% hydrogen peroxide in water solution and acetonitrile (8 ml) were introduced into the reactor. The reactor heated to 150 °C within 30 min with stirring, and the reactor was held at this temperature for 2 h. Oxidations of substituted aromatics over CNB0.15 using O2 as oxidant were also carried out in a Teflon-lined stainless steel batch reactor. 1 mmol Substrate, 4 ml acetonitrile, 50 mg CNB0.15 were loaded into the reactor. The reactor was sealed, purged with O2 to remove the air for 3 times, increased the O2 pressure to 1 MPa, and then the reactor was heated to the desired temperature. After reaction the reactor was placed in ice water to quench the reaction and the products were analyzed with a GC and GC-MS (Agilent Technologies, GC6890N, MS5970) with benzyl chloride as the internal standard.Reusing of the catalysts
For the recycling experiments, the reaction mixture was centrifuged for 10 min after the first reaction and the liquid layer was siphoned out. The residual solid was washed with anhydrous ethanol and centrifuged twice and finally dried in a vacuum oven at 80 °C for 6 h. Aromatics, H2O2 and acetonitrile were then charged for subsequent round of reaction under otherwise identical conditions.GC and GC-MS analysis
All GC experiments were carried out and recorded using a HP 6890 Series. The GC conditions for the analysis of the aromatic oxidation products were as follows. Injection port temperature 280 °C, column temperature: initial temperature: 50 °C (1 min); Gradient rate: 50 °C (4 min); Final temperature 250 °C (5 min); Flow rate: 80 ml min−1. All GC-MS experiments were carried out and recorded using an Agilent GC6890N, MS5970 series. The GC-MS conditions for the products analysis was kept the same as that used on GC. Selectivities and conversions were calculated from the equations: Selectivity = [aldehyde or ketone]/[Consumed aromatics] ×100; Conversion = [Consumed aromatics]/[Initial aromatics] × 100, respectively.Results and discussion
The structures of these hybrid materials were first investigated by powder X-ray diffraction (XRD). The XRD pattern (Fig. 1a) supports the preservation of the graphite-like packing of practically all products, showing the typical (002) interlayer-stacking peak around 27.4°, as is well known for the melon network.39,50 The FTIR spectra (Fig. S1 ESI†) of the CNB samples showed typical C–N heterocycle stretches in the region of ca. 1100–1600 cm−1 and the breathing mode of the tri-s-triazine units at 800 cm−1 evidencing that the original graphitic C–N network remained. The typical vibration of B–N at 1370 cm−1 is presumably overlapped by that of the C–N stretch.51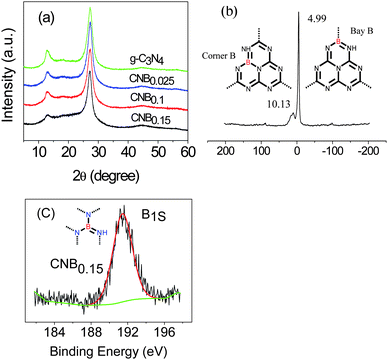 | ||
| Fig. 1 (a) XRD patterns of the B doped and pristine g-C3N4; (b) 11B solid-state MAS NMR spectra of CNB0.15, inset: the building unit of C3N4 with B doping at the bay-carbon site (right) and at the corner-carbon site (left), the dotted lines imply that the tricyclic system is a repeat unit in a regular lattice framework; (c) XPS spectra of CNB0.15. | ||
Structural details on the incorporation of boron into the C/N matrix were obtained with 11B solid-state MAS NMR experiment. Fig. 1b suggested two peaks, one strong peak at 4.99 ppm and one small peak at 10.13 ppm, which corresponded to two different positions in the framework structure, say as the bay-carbon site and the corner carbon site (Fig. 1b, inset). Further evidence of the boron doping came from the X-ray photoelectron spectroscopy (XPS) measurements. The XPS result gives direct evidence of a boron heteroatom in the doped g-C3N4 as well as the degree of doping (Fig. 1c).The B1s binding energy (BE) peaks of CNB0.15 is centered at ca. 191.6 eV, which is at a higher binding energy than that for h-BN (190.0 eV) and at a lower binding energy than that for Kawaguchi's BCN(H) (192.1 eV, boron surrounded by three nitrogen atoms), which indicates that the boron atoms in our CNB network are more electropositive than that in h-BN, but are not as electropositive as that in Kawaguchi's BCN(H).52 The boron content (CNB0.15) as determined by XPS is near 9 mol%, i.e. a tentative sum formula of the structure is C3N4.7B0.8H1.9.
UV-Vis spectra (Fig. S2 ESI†) indicate that the optical band gap and thereby the semiconductor properties of the CNB have indeed changed, with an extension of the spectrum towards absorption in the visible range. This can in principle enhance the ability to harvest visible light, beneficial for heterogeneous photocatalysis (Table S1 ESI†), which is however not the subject of the present work. It is also to be said that the incorporation of boron is expected to lower the HOMO position of the resulting material, thus increasing the potential oxidation strength as compared to bare carbon nitrides.
Catalytic testing was first performed for the oxidation of toluene using CNB with varying boron content as metal-free catalysts. The corresponding conversions are summarized in Table 1. In all the cases, 2 to 6.3% conversions were found, while the blank run without catalyst gave almost no product; that is, CNB (including the non doped g-C3N4) indeed acts as an oxidation catalyst. The doping of boron atoms in the “melon” texture undoubtedly increased the activity. The g-C3N4 itself gave minor conversion of 2%, while all the boron doped materials appeared to be more active as illustrated in Table 1. Among all the catalysts tested, CNB0.15 gave the best conversion of up to 6.3%. The catalysts are highly stable and can be reused for several cycles without loss of activity (Table 1, entry 5, 6, 7), which is a prerequisite for practical applications. Also this fact however proves that the amount of reaction is presumably limited by the availability of an oxidation agent, only. For practical applications, this would be ensured by a constant influx of H2O2, which however cannot be performed in our autoclave setup. Increasing the ratio of ntoluene/nH2O2 to 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5, yielded a 12.4% conversion of toluene (Table 1, entry 9). GC-MS analyses of the post-reaction mixture showed that the CNB catalysts (beside CNB0.25) afford remarkably high selectivities towards the formation of benzaldehyde, i.e. almost all the oxidation reaction gave a selectivity above 99%. It should be noted, that the activity of CNB did not simply increase with the amount of boron doped, as exceeding 9 mol% appears to be detrimental (Table S2 ESI†).
:5, yielded a 12.4% conversion of toluene (Table 1, entry 9). GC-MS analyses of the post-reaction mixture showed that the CNB catalysts (beside CNB0.25) afford remarkably high selectivities towards the formation of benzaldehyde, i.e. almost all the oxidation reaction gave a selectivity above 99%. It should be noted, that the activity of CNB did not simply increase with the amount of boron doped, as exceeding 9 mol% appears to be detrimental (Table S2 ESI†).
| Entry | Catalyst | Conv. [%] | Sel. [%] BAb |
|---|---|---|---|
| a Reaction conditions: toluene 0.8 ml, H2O2 (30% in water solution) 0.77 ml, catalyst 50 mg, acetonitrile 8 ml, reaction temperature 150 °C, reaction time 2 h. b BA = benzaldehyde. c Second run to test the reusability of catalyst. d Third run to test the reusability of the catalyst. e Forth run to test the reusability of the catalyst. f Mole ratio nH2O2/ntoluene = 3. g Mole ratio nH2O2/ntoluene = 5. h Mole ratio nH2O2/ntoluene = 7. | |||
| 1 | g-C3N4 | 2.0 | >99.0 |
| 2 | CNB0.025 | 4.2 | >99.0 |
| 3 | CNB0.1 | 5.9 | >99.0 |
| 4 | CNB0.15 | 6.3 | >99.0 |
| 5c | CNB0.15 | 6.2 | >99.0 |
| 6d | CNB0.15 | 6.1 | >99.0 |
| 7e | CNB0.15 | 6.3 | >99.0 |
| 8f | CNB0.15 | 9.8 | >99.0 |
| 9g | CNB0.15 | 12.4 | >99.0 |
| 10h | CNB0.15 | 11.5 | >99.0 |
The C–H bond energy of the aliphatic toluene bond is 90 kcal mol−1, while the corresponding C–H bond of ethylbenzene has 87 kcal mol−1.53 This should enable similar organocatalytic oxidation chemistry of ethylbenzene towards acetophenone. In fact, using H2O2 as the oxidant and keeping T = 150 °C for 2 h, GC-MS analysis indeed disclosed the formation of acetophenone in ∼4.6% and 10.6% yield, respectively, based on the CNBx species (see Table 2), with the same remarkable selectivities exceeding 99%.
| Entry | Catalyst | Conv. [%] | Sel. [%] APb |
|---|---|---|---|
| a Reaction conditions: ethylbenzene 0.8 ml, H2O2 (30% in water solution) 0.66 ml, catalyst 50 mg, acetonitrile 8 ml, reaction temperature 150 °C, reaction time 2 h. b AP = acetophenone. | |||
| 1 | g-C3N4 | 4.6 | >99.0 |
| 2 | CNB0.025 | 5.9 | >99.0 |
| 3 | CNB0.1 | 7.4 | >99.0 |
| 4 | CNB0.15 | 10.6 | >99.0 |
With the preliminary success in the oxidation of toluene and ethylbenzene using H2O2 as oxidant, we also addressed the oxidation of substituted benzylic aromatics using molecular oxygen (O2) an oxidant which is much harder to activate and tame. Since CNB0.15 was found to be the best catalyst for the oxidation of toluene and ethylbenzene, it was used in all further experiments, the results of which are summarized in Table 3.
| Entry | Substrate | Main product | BDE b | T (°C) | Conv. (%) | Sel. (%) |
|---|---|---|---|---|---|---|
| a Reaction conditions: substrates 1 mmol, O2 pressure 1 MPa, catalyst 50 mg, acetonitrile 4 ml, reaction time 24 h. b C–H bond dissociation energy from the literature.54 c Using g-C3N4 as catalyst. | ||||||
| 1 |
|
|
81 | 160 | 20.3 | 95.1 |
| 2 |
|
|
80 | 130 | 45.7 | >99.0 |
| 3c |
|
|
80 | 130 | 28.8 | >99.0 |
| 4 |
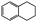
|
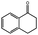
|
83 | 130 | 72.2 | 87.5 |
| 5 |
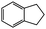
|
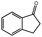
|
82 | 115 | 36.7 | >99.0 |
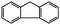
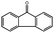
Although no products had been detected in the case of toluene and ethylbenzene using O2 as the oxidant, oxidation of other substrates with an activated benzylic position went smoothly. Using oxidation of fluorene (C–H binding energy:54 80 kcal mol−1) to fluorenone as an example, 45.7% conversion and 99% selectivity to fluorenone were achieved in 24 h at 130 °C using 1 MPa O2 pressure. As a control experiment, by simply heating the same reaction mixture without the adding of catalyst, almost no fluorenone could be detected by GC-MS.
Conclusions
To conclude, we have presented a new synthesis of boron doped carbon nitrides, using BH3NH3 as the boron source. The catalytic properties of these materials were tested in the oxidation of activated aliphatic C–H bonds. These materials exhibit good catalytic performance for the selective oxidation of toluene and substituted benzylic aromatics. Compared to traditional metal catalysts, the metal-free, boron doped, organic semiconducting polymers afford a remarkably high selectivity towards the formation of aldehydes or ketones. In addition they are highly stable and could be recycled several times without losing their reactivity. The range of substrates depends on the doping of the catalyst as well as on the oxidation agent applied. Using H2O2, the best catalyst of this series is already able to oxidize methylaromatic systems, while with molecular oxygen, the reaction is restricted to more activated methylene groups. The catalysts are therefore rather selective, with respect to both the substrate and the product they create. This gives hope that further modifications of similar catalytically active polymers and solid state materials will allow access to other more demanding substrates such as alkanes or even methane (C–H bond energy:54 105 kcal mol−1) while keeping the selectivity. It is hoped that our findings will stimulate such further investigations, both on the materials as well as on the applications.Acknowledgements
This work was supported by ENERCHEM project of MPI and the National Science Foundation of China (No. 20806065 and No. 20990221). We thank Dr Yan Di for testing the hydrogen evolution reactions.Notes and references
- M. Christmann, Angew. Chem., Int. Ed., 2008, 47, 2740–2742 CrossRef.
- G. Dyker, Angew. Chem., Int. Ed., 1999, 38, 1698–1712 CrossRef.
- C. G. Jia, T. Kitamura and Y. Fujiwara, Acc. Chem. Res., 2001, 34, 633–639 CrossRef CAS.
- A. E. Shilov and G. B. Shul'pin, Chem. Rev., 1997, 97, 2879–2932 CrossRef CAS.
- J. Zhang, X. Liu, R. Blume, A. H. Zhang, R. Schlogl and D. S. Su, Science, 2008, 322, 73–77 CrossRef CAS.
- K. Kamata, K. Yonehara, Y. Nakagawa, K. Uehara and N. Mizuno, Nat. Chem., 2010, 2, 478–483 CrossRef CAS.
- M. S. Chen and M. C. White, Science, 2010, 327, 566–571 CrossRef CAS.
- Y. Wang, J. S. Zhang, X. C. Wang, M. Antonietti and H. R. Li, Angew. Chem., Int. Ed., 2010, 49, 3356–3359 CAS.
- J. A. Labinger and J. E. Bercaw, Nature, 2002, 417, 507–514 CrossRef CAS.
- T. Punniyamurthy, S. Velusamy and J. Iqbal, Chem. Rev., 2005, 105, 2329–2363 CrossRef CAS.
- F. Rajabi, J. H. Clark, B. Karimi and D. J. Macquarrie, Org. Biomol. Chem., 2005, 3, 725–726 RSC.
- S. M. Yiu, Z. B. Wu, C. K. Mak and T. C. Lau, J. Am. Chem. Soc., 2004, 126, 14921–14929 CrossRef CAS.
- D. R. Dreyer, H.-P. Jia and C. W. Bielawski, Angew. Chem., Int. Ed., 2010, 49, 6686–6816 CrossRef.
- F. Wang, J. Xu, X. Q. Li, J. Gao, L. P. Zhou and R. Ohnishi, Adv. Synth. Catal., 2005, 347, 1987–1992 CrossRef CAS.
- F. Konietzni, U. Kolb, U. Dingerdissen and W. F. Maier, J. Catal., 1998, 176, 527–535 CrossRef CAS.
- S. Das, C. D. Incarvito, R. H. Crabtree and G. W. Brudvig, Science, 2006, 312, 1941–1943 CrossRef CAS.
- L. Que and W. B. Tolman, Nature, 2008, 455, 333–340 CrossRef.
- M. Yamashita, H. Furutachi, T. Tosha, S. Fujinami, W. Saito, Y. Maeda, K. Takahashi, K. Tanaka, T. Kitagawa and M. Suzuki, J. Am. Chem. Soc., 2007, 129, 2–3 CrossRef CAS.
- B. J. Wallar and J. D. Lipscomb, Chem. Rev., 1996, 96, 2625–2657 CrossRef CAS.
- J. Valton, M. Fontecave, T. Douki, S. G. Kendrew and V. Niviere, J. Biol. Chem., 2005, 281, 27–35 CrossRef.
- I. Siewert, C. Limberg, S. Demeshko and E. Hoppe, Chem.–Eur. J., 2008, 14, 9377–9388 CrossRef CAS.
- C. Limberg, Angew. Chem., Int. Ed., 2003, 42, 5932–5954 CrossRef CAS.
- M. Fontecave, S. Menage and C. Duboc-Toia, Coord. Chem. Rev., 1998, 178–180, 1555–1572 CrossRef CAS.
- M. W. Grinstaff, M. G. Hill, J. A. Labinger and H. B. Gray, Science, 1994, 264, 1311–1313 CAS.
- F. Bedioui, Coord. Chem. Rev., 1995, 144, 39–68 CrossRef CAS.
- M. H. So, Y. G. Liu, C. M. Ho and C. M. Che, Chem.–Asian J., 2009, 4, 1551–1561 CrossRef CAS.
- D. Mansuy, Coord. Chem. Rev., 1993, 125, 129–141 CrossRef CAS.
- M. Costas, M. P. Mehn, M. P. Jensen and L. Que, Chem. Rev., 2004, 104, 939–986 CrossRef CAS.
- R. F. Parton, I. F. J. Vankelecom, M. J. A. Casselman, C. P. Bezoukhanova, J. B. Uytterhoeven and P. A. Jacobs, Nature, 1994, 370, 541–544 CrossRef CAS.
- I. Siewert and C. Limberg, Chem.–Eur. J., 2009, 15, 10316–10328 CrossRef CAS.
- N. Kitajima, M. Ito, H. Fukui and Y. Morooka, J. Chem. Soc., Chem. Commun., 1991, 102–104 RSC.
- Z. H. Zhang, K. Leinenweber, M. Bauer, L. A. J. Garvie, P. F. McMillan and G. H. Wolf, J. Am. Chem. Soc., 2001, 123, 7788–7796 CrossRef CAS.
- Y. Wang, X. C. Wang, M. Antonietti and Y. J. Zhang, ChemSusChem, 2010, 3, 435–439 CrossRef CAS.
- T. Komatsu, J. Mater. Chem., 2001, 11, 802–805 RSC.
- J. Gracia and P. Kroll, J. Mater. Chem., 2009, 19, 3013–3019 RSC.
- J. R. Holst and E. G. Gillan, J. Am. Chem. Soc., 2008, 130, 7373–7379 CrossRef CAS.
- J. L. Zimmerman, R. Williams, V. N. Khabashesku and J. L. Margrave, Nano Lett., 2001, 1, 731–734 CrossRef CAS.
- M. Antonietti and P. Fratzl, Macromol. Chem. Phys., 2010, 211, 166–170 CrossRef CAS.
- A. Thomas, A. Fischer, F. Goettmann, M. Antonietti, J. O. Muller, R. Schlogl and J. M. Carlsson, J. Mater. Chem., 2008, 18, 4893–4908 RSC.
- X. Jin, V. V. Balasubramanian, S. T. Selvan, D. P. Sawant, M. A. Chari, G. Q. Lu and A. Vinu, Angew. Chem., Int. Ed., 2009, 48, 7884–7887 CrossRef CAS.
- J. Zhu, Y. Wei, W. Chen, Z. Zhao and A. Thomas, Chem. Commun., 2010, 46, 6965–6967 RSC.
- Y. Wang, Y. Di, M. Antonietti, H. Li, X. Chen and X. Wang, Chem. Mater., 2010, 22, 5119–5121 CrossRef CAS.
- S. C. Yan, Z. S. Li and Z. G. Zou, Langmuir, 2010, 26, 3894–3901 CrossRef CAS.
- G. Liu, P. Niu, C. Sun, S. C. Smith, Z. Chen, G. Q. Lu and H.-M. Cheng, J. Am. Chem. Soc., 2010, 132, 11642–11648 CrossRef CAS.
- Y. J. Zhang, T. Mori, J. H. Ye and M. Antonietti, J. Am. Chem. Soc., 2010, 132, 6294–6295 CrossRef CAS.
- Z. J. Li, G. D. Del Cul, W. F. Yan, C. D. Liang and S. Dai, J. Am. Chem. Soc., 2004, 126, 12782–12783 CrossRef CAS.
- S. In, A. Orlov, R. Berg, F. Garcia, S. Pedrosa-Jimenez, M. S. Tikhov, D. S. Wright and R. M. Lambert, J. Am. Chem. Soc., 2007, 129, 13790–13791 CrossRef CAS.
- O. Stephan, P. M. Ajayan, C. Colliex, P. Redlich, J. M. Lambert, P. Bernier and P. Lefin, Science, 1994, 266, 1683–1685 CAS.
- W. L. Wang, X. D. Bai, K. H. Liu, Z. Xu, D. Golberg, Y. Bando and E. G. Wang, J. Am. Chem. Soc., 2006, 128, 6530–6531 CrossRef CAS.
- T. Komatsu and T. Nakamura, J. Mater. Chem., 2001, 11, 474–478 RSC.
- A. Y. Liu, R. M. Wentzcovitch and M. L. Cohen, Phys. Rev. B: Condens. Matter, 1989, 39, 1760–1765 CrossRef CAS.
- M. Kawaguchi, T. Kawashima and T. Nakajima, Chem. Mater., 1996, 8, 1197–1201 CrossRef CAS.
- H. R. Lucas, L. Li, A. A. N. Sarjeant, M. A. Vance, E. I. Solomon and K. D. Karlin, J. Am. Chem. Soc., 2009, 131, 3230–3245 CrossRef CAS.
- L. J. J. Laarhoven and P. Mulder, J. Phys. Chem. B, 1997, 101, 73–77 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Additional analytical and experimental data. See DOI: 10.1039/c0sc00475h |
| This journal is © The Royal Society of Chemistry 2011 |