A dual-functional tetrakis-imidazolium macrocycle for supramolecular assembly†
Christopher J.
Serpell
a,
James
Cookson
b,
Amber L.
Thompson
a and
Paul D.
Beer
*a
aChemistry Research Laboratory, Department of Chemistry, University of Oxford, Mansfield Road, Oxford, OX1 3TA, United Kingdom. E-mail: paul.beer@chem.ox.ac.uk; Fax: +44 (0) 1865 272609; Tel: +44 (0) 1865 285142
bJohnson Matthey Technology Centre, Blount's Court, Sonning Common, Reading, RG4 9NH, UK. E-mail: cooksj@matthey.com; Fax: +44(0) 118 924 2338; Tel: +44 (0) 118 924 2061
First published on 17th December 2010
Abstract
A new versatile tetrakis-imidazolium macrocycle for use in supramolecular applications is reported. It displays excellent affinities for π-electron rich neutral guests such as 1,5-dihydroxynaphthalene derivatives and TTF, providing opportunities for the construction of interlocked molecules, as well as exhibiting extensive and potent anion coordination chemistry.
Introduction
The tetra-cationic viologen macrocycle cyclobis(paraquat-p-phenylene) (CBPQT4+, Fig. 1), also known as the ‘blue box,’ popularised by Stoddart and co-workers has been extensively used in supramolecular assembly in recent years, stemming from its ability to form charge-transfer complexes with electron rich species. These assemblies are often coloured1,2 and are formed promiscuously with guests such as hydroquinone, benzidine,3 indole,4 tetrathiafulvalene (TTF),5 amino acids,6 and neurotransmitters.7 The guests are bound with well-defined preferences, permitting the construction of switchable multi-station rotaxanes and catenanes,8 and further incorporation into mechanised nanoparticles,9 molecular electronic devices,10 and metal–organic frameworks.11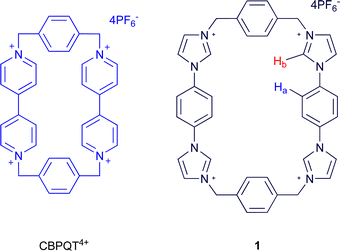 | ||
| Fig. 1 CBPQT4+ and macrocycle 1 | ||
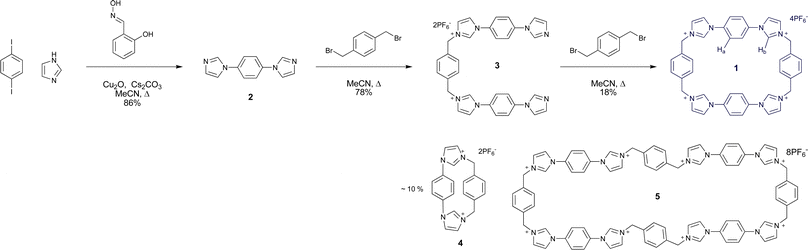 | ||
| Scheme 1 Synthesis of macrocycle 1 | ||
Despite these impressive achievements, there are some less well-publicised drawbacks to this compound concerning its stability—it is vulnerable to decomposition at the hands of reducing agents, bases, and nucleophiles.12 There is therefore still room for improvement upon the CBPQT4+ system. A number of strategies have been pursued in the development of CBPQT4+: it may be covalently tethered to other components,13 isomerically varied at the phenylene unit,14 or expanded using a biphenylene spacer.15 Alternatively, the phenylene unit can be replaced by a chiral16 or photochemically switchable moiety.17 In our own research, substitution of one phenylene spacer with an isophthalamide motif was achieved, permitting the construction of an indolocarbazole [2]rotaxane host system which exhibited chloride anion selectivity.18 However, none of these studies addressed the underlying instability of the system which arises from ring strain and the redox activity of the paraquat moieties.
Recent interest has therefore turned towards the construction of more stable alternatives to CBPQT4+. During the course of the research presented herein, Sessler and co-workers reported a tetrakis-imidazolium macrocycle as a larger alternative to CBPQT4+.19 Although this was found to form chain-like inclusion complexes with the mono-terephthalate anion, and silver carbene cage systems,20 no further applications of this system have yet been reported.
Herein we report the synthesis of a new tetrakis-imidazolium macrocycle 1 (Fig. 1) capable of forming strong complexes with a range of both neutral π-electron rich species and anionic guests. By making use of the 1,4-diimidazolium-benzene unit rather than 4,4′-bipyridinium, the ring strain can be relieved, thus reducing its susceptibility to nucleophilic attack relative to CBPQT4+. Moreover, the larger macrocycle annulus alters the selectivity for π-electron rich guests, and together with anion recognition properties via charge-assisted C–H hydrogen bonds, the new macrocycle could be potentially exploited in the design of future molecular machines.
Results and discussion
Synthesis
Macrocycle 1 was synthesised according to Scheme 1.21 Firstly, hetero-Ullmann coupling was used to synthesise 1,4-diimidazolyl benzene 2 from 1,4-diiodobenzene and imidazole,22 giving 2 as a yellow solid in 86% yield. Reaction of α,α′-dibromo-p-xylene with 2 in acetonitrile gave a bromide salt precipitate, which upon anion exchange produced the organic solvent soluble hexafluorophosphate salt 3 in 78% yield.Macrocyclisation was achieved by high dilution reaction of 3 and α,α′-dibromo-p-xylene in refluxing acetonitrile. The product was separated by selective precipitation and subjected to anion exchange, giving 1 as a white solid in 18% yield. This compound was found to be soluble in polar organic solvents such as acetone and acetonitrile.
Side products were also produced during the final cyclisation step. The contracted bis-imidazolium macrocycle 4 was identified as the major side product (ca. 10% yield), giving a characteristically distinct 1H NMR spectrum in acetone-d6 and a singly charged HR-ESMS peak at m/z = 313.1443 corresponding to [M–H]+. The generation of 4 indicates that the N-alkylation reaction is reversible, since one of these bonds must be broken to form the small macrocycle.
Slow diffusion of chloroform vapour into an acetone solution of the mixture containing the side products gave crystals suitable for structural analysis using synchrotron X-ray radiation at Diamond Light Source beamline I19, revealing the unprecedented octa-imidazolium macrocycle 5 with a remarkable end-to-end distance of 28 Å (Fig. 2a). While this is clearly a very interesting species, it comprised only a small fraction of the bulk material, and so could not be studied further. Investigations are underway into the optimisation of the synthesis in favour of either 1 or 5 using templating agents.23
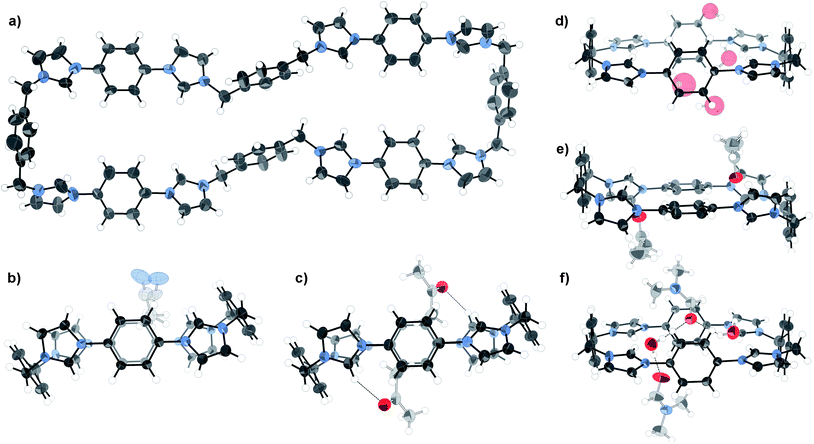 | ||
| Fig. 2 X-ray crystal structures of a) octa-imidazolium macrocycle 5 (omitting anions, solvent, and minor components of disorder for clarity), and b) acetonitrile, c) acetone, d) water, e) acetone) and f) DMF/water solvates of 1 (omitting anions and selected solvent for clarity). Thermal ellipsoids displayed at 50% probability. | ||
Five distinct crystal structures24–29 have been obtained for the tetra-cationic macrocycle 1 itself when crystallised from different solvents (Fig. 2b–f), giving a measure of its flexibility and supramolecular potential. Two conformational modes were observed—a picket fence-type structure (Fig. 2b–c) with the acidic imidazolium hydrogen bond donors (Hb) pointing perpendicularly to the plane of the molecule, suitable for the inclusion of π-electron rich guests, and a flattened arrangement with convergent hydrogen bond donors (Fig. 2d–f), ideal for association with anions.
The picket-fence structures are almost conformationally identical with respect to the macrocycle. Their packing motifs contrast, however, such that while the acetonitrile solvate forms layers with the macrocycle in the same orientation, the acetone analogue involves alternating perpendicularly rotated chains of the tetra-cation. Comparison of the flattened structures reveals that the water/DMF and water solvates have very similar conformations, with the phenylene units skewed with respect to the plane of the macrocycle. Indeed, examination of the packing reveals that the structures are isomorphous with respect to the macrocycle. In contrast, the second acetone solvate structure (Fig. 2e) displays the phenylene groups in the plane of the molecule with canted imidazolium units.
Comparison of these crystal structures with that of CBPQT4+ confirms that the cavity of 1 is longer (12.0–12.5 Å versus 10.8 Å), while the relief of ring strain is manifested by the increased width of the annulus (7.5–8.1 Å versus 6.1 Å).23
Association with electron rich neutral guests
The propensity of macrocycle 1 to form inclusion complexes with π-electron rich neutral guests (Fig. 3) was probed using 1H NMR titrations and crystallographic investigations.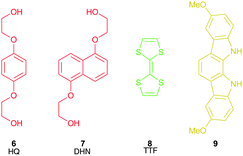 | ||
| Fig. 3 Electron rich neutral guests. | ||
Alkyoxy-arenes such as hydroquinone derivatives are electron rich, and are known to form complexes with CBPQT4+. In particular, 1,5-dihydroxynaphthalene (DHN) derivatives have been widely used as a station in molecular shuttles.8,13,301H NMR titration experiments were conducted in acetonitrile-d3 with the bis-(2-hydroxyethyl) ethers of hydroquinone (HQ) and 1,5-dihydroxynaphthalene (6 and 7 respectively, Fig. 3). Addition of the naphthalene compound 7 resulted in a significant upfield perturbation of the macrocycle phenylene (Ha) proton (Δδ10eqv = −0.477 ppm). Job plot analysis of the titration data and curve fitting using WinEQNMR231 yielded a two-stage equilibrium with K1 = 1880 mol−1 dm3 and K2 = 52 mol−1 dm3. The first association constant is large with respect to the second, indicating that a [2]pseudorotaxane complex can be proposed as the major species in solution. No association was observed with the hydroquinone derivative 6 in this solvent.
TTF (8, Fig. 3) is also commonly employed as a station in shuttling rotaxanes and catenanes. TTF readily forms π-complexes with electron deficient neutral molecules32 and cationic species,33 and can be reversibly oxidised by mild reagents.34 In its neutral form TTF is more strongly bound to CBPQT4+ than dialkoxynaphthalenes and is thus the preferred site in bistable rotaxanes and catenanes. Titration of macrocycle 1 with TTF in acetonitrile-d3 resulted in modest upfield shifts of the phenylene resonance, but a more significant downfield purturbation of the Hb peak (Δδ10eqv = +0.100 ppm), giving an association constant of 752 (120) mol−1 dm3. In contrast to CBPQT4+ (which binds TTF with a Ka > 104 mol−1 dm3 in the same solvent35), macrocycle 1 displays more than twice the binding affinity for 1,5-dihydroxynaphthalene systems than TTF.
Indolo[2,3-a]carbazoles are extended aromatic species containing two pyrrole units presenting convergent hydrogen bond donors. Their π-electron rich nature facilitates the formation of charge-transfer complexes with paraquat derivatives,18 while their acidic protons can be used to bind anions.36–43
1H NMR titration experiments using dimethoxy-indolocarbazole 9 (Fig. 3) and macrocycle 1 in acetonitrile-d3 were hampered by the poor solubility of 9. However, a significant upfield perturbation (Δδ = −0.248 ppm) of the phenylene Ha proton peak was observed for the addition of three equivalents of 9 to a solution of 1, consistent with a π–π stacked complex. Job plot analysis of the titration data could not unambiguously confirm a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 or 1:2 stoichiometry. The nature of the association was revealed when an X-ray crystal structure was obtained of the co-crystal formed by 1 and 9 (Fig. 4). It was found that the indolocarbazole associates with the macrocycle, but that it is bound externally, maximising the π-overlap between the two components. This arrangement permits the formation of 1:2, 1:1, or 2:1 complexes, explaining the stoichiometric ambiguity.
:1 or 1:2 stoichiometry. The nature of the association was revealed when an X-ray crystal structure was obtained of the co-crystal formed by 1 and 9 (Fig. 4). It was found that the indolocarbazole associates with the macrocycle, but that it is bound externally, maximising the π-overlap between the two components. This arrangement permits the formation of 1:2, 1:1, or 2:1 complexes, explaining the stoichiometric ambiguity.
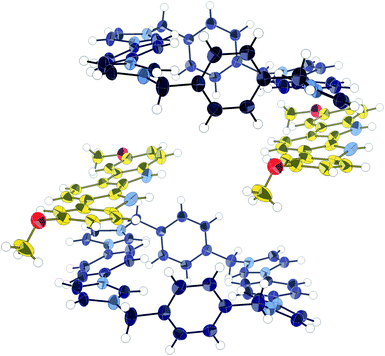 | ||
| Fig. 4 X-ray crystal structure of 1·9 co-crystal. Thermal ellipsoids displayed at 50% probability. Anions, solvent, and disorder omitted for clarity. | ||
Towards rotaxane formation
Given the strength of association exhibited by macrocycle 1 with 7, the focus shifted towards the construction of interlocked systems. A 1,5-dialkoxynaphthalene rotaxane was therefore envisaged using a new stoppering methodology (Scheme 2). The addition of dicobalt octacarbonyl across triple bonds occurs rapidly and quantitatively,44,45 thus appending significant steric bulk in a highly facile manner. This transformation is therefore an attractive stoppering strategy for the construction of interlocked structures.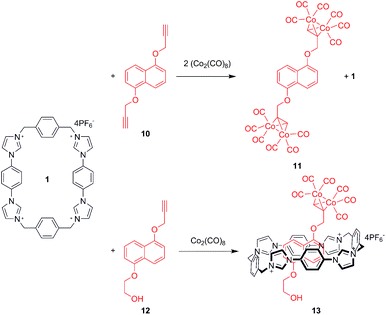
Initial 1H NMR pseudorotaxane experiments were conducted in order to verify that the cobalt carbonyl group possessed sufficient steric bulk to prevent dethreading (Supplementary Info Fig S3†). Significant perturbations of the Ha proton resonance occurred upon addition of 10 to 1 in acetone. Conversely, the addition of cobalt carbonyl complex stoppered thread 11 caused no movement in the 1H NMR signals of 1 which confirms that the organometallic stoppers inhibit interpenetrative slippage.
Addition of dicobalt octacarbonyl to an acetone solution of 1 and 12 in acetone, followed by evaporation of the solvent produced large red crystals which were subjected to X-ray structural analysis, revealing pseudorotaxane 13. The structure (Fig. 5) was found to consist of the cobalt-complexed thread sitting within the macrocycle cavity, participating in charge assisted π–π stacking between the naphthalene unit and the bis-imidazolium-phenylene portion of the macrocycle as expected. The thread was disordered such that it could reside equally as displayed or rotated 180° about the axis passing through the centre of the naphthalene and macrocycle phenylene units. The same form of disorder has been observed in the solid state complex of CBPQT4+ with catechol dimethyl ether.46 Importantly, the carbonyl groups extended beyond the edge of the macrocycle, confirming the efficacy of the cobalt carbonyl group as a sufficiently bulky stopper for this macrocycle.
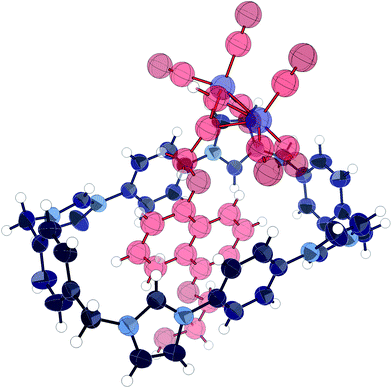 | ||
| Fig. 5 X-ray crystal structure of pseudorotaxane 13. Thermal ellipsoids displayed at 50% probability. Anions, solvent, and disorder omitted for clarity. | ||
Addition of excess dicobalt octacarbonyl to an equimolar solution of 1 and 1,5-dihydroxynaphthalene bis-propargyl ether (10) in acetone (Scheme 2) appeared to produce a highly polar red substance (when analysed by thin layer chromatography) consistent with formation of the target rotaxane. Disappointingly, this was found to decompose spontaneously and could not therefore be isolated (despite the stoppered thread 11 being obtainable). This instability may be attributable to unfavourable steric interactions, to which alkyne-dicobalt hexacarbonyl complexes are vulnerable.47 Further variation of components and reaction conditions was hampered by the polar/apolar mis-match of 1 and dicobalt octacarbonyl, limiting the range of solvents available.
Although isolation of a dicobalt hexacarbonyl stoppered naphthalene rotaxane was ultimately unsuccessful, the crystal structure of 13 does confirm the threading of naphthalene components through 1, and cobalt carbonyl complexes have been introduced as a new stoppering methodology. Macrocycle 1 is thus shown to form complexes with dihydroxynaphthalene derivatives (bound internally), TTF, and indolocarbazole (bound externally). In contrast to CBPQT4+, the DHN thread is held more strongly than TTF. Importantly, no evidence of macrocycle degradation was seen under any of the conditions used.
Association with anionic guests
The tetrakis-imidazolium macrocycle 1 posesses four C–H hydrogen bond donors in addition to its positive charge, and is thus a potentially potent host for anionic guests (Fig. 6). A number of tetrakis-imidazolium anion receptors have been reported, which display geometrically controlled selectivities for species such as fluoride,48,49 chloride,50 bromide,51 dicarboxylates,52 and sulfate,53 although few are macrocyclic.48,49,51 The conformational flexibility of macrocycle 1 could permit the binding of anions displaying a range of geometries.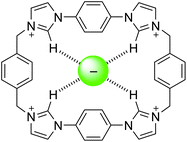 | ||
| Fig. 6 Hydrogen bond mediated anion coordination by 1. | ||
To delineate the anion binding properties of 1, 1H NMR titrations were conducted in DMSO-d6. Crystallographic analysis was performed where possible, giving a descriptive evaluation of the anion binding environment in the solid state.
In general, addition of anions as their tetrabutylammonium (TBA) salts resulted in significant downfield perturbations of the macrocycle's acidic imidazolium Hb1H NMR resonance, and minor changes in the other peaks. Job plot analysis of the titration data was performed to establish the binding stoichiometry (1:1 unless stated otherwise), and curve-fitting was used to determine association constants. The results are summarised in Table 1.
:1 host:guest model) and 1H NMR peak perturbations for the interaction of anions with macrocycle 1 in DMSO-d6. Measured at 293 K, macrocycle concentration 2 × 10−3 mol dm−3. Errors given in brackets
| Anion | Ka/mol−1 dm3 | Hb Δδ10eqv/ppm |
|---|---|---|
| a Precipitated after addition of four equivalents of anion. b Three equivalents. c Negligible binding strength. d Stoichiometrically ambiguous. | ||
| Cl− | 420 (23) | +0.514 |
| Br− | 241 (3) | +0.214 |
| I− | 120 (1) | +0.058 |
| H2PO4− | +0.061b | |
| HSO4− | 102 (7) | +0.053 |
| ClO4− | −0.027 | |
| N3− | 488 (46) | +0.069 |
| SCN− | ||
| AcO− | 467 (23) | +1.130 |
| BzO− | 359 (9)d | +0.840 |
| AuCl2− | 245 (4) | −0.174 |
| Pd2Cl62− | 1219 (26) | +0.155 |
The association constant for chloride binding was found to be twice that of bromide, whereas bromide gave an association constant twice that of iodide. The strength of halide binding reflects the basicity series, but size selectivity may also play a role. The basic anion fluoride induced deprotonation, as manifested in a pronounced broadening and loss of symmetry in the 1H NMR spectrum, and the development of a yellow colour in the solution. A crystal structure of the tetrachloride salt of 1 was obtained (Fig. 7a), revealing the macrocycle in the picket-fence conformation, creating channels filled with disordered DMF (omitted for clarity in Fig. 7a). Although this conformation is not consistent with a convergent hydrogen bond model suggested by the 1:1 binding stoichiometry, and the large shift for the acidic imidazolium Hb proton NMR resonance observed in solution, it should be noted that, in addition to the change of stoichiometry, solution and solid-phase conditions are very different: in crystal packing it is essential that strong hydrogen bond donor–acceptor interactions are maximised, a condition best satisfied in a divergent array.
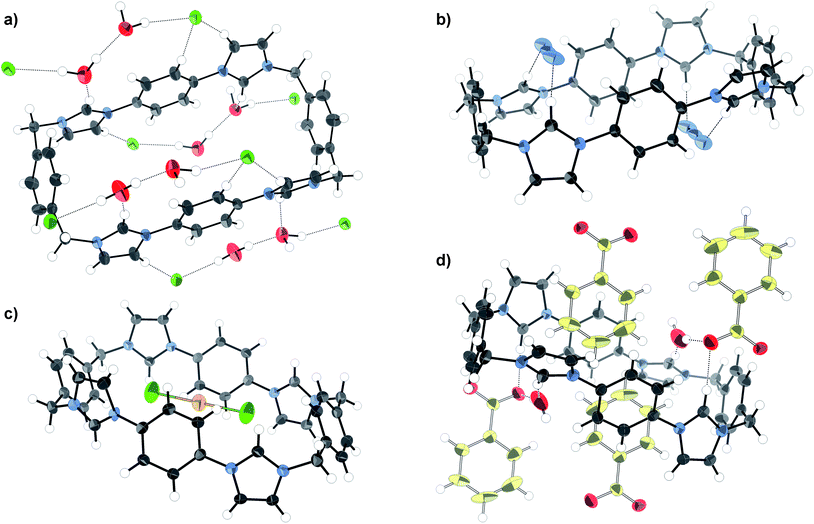 | ||
| Fig. 7 X-ray crystal structures of the a) chloride, b) azide, c) AuCl2− and d) benzoate salts of macrocycle 1. Thermal ellipsoids displayed at 50% probability, selected solvent omitted for clarity. | ||
The effects of the basicity trend could also be observed in the singly charged oxoanions H2PO4−, HSO4−, and ClO4−. Importantly, these examples are all of tetrahedral shape, and so offer a chance to study the interplay of basicity and geometry when compared with the halides. Association of 1 with H2PO4− was manifested by total precipitation after the addition of four equivalents of the anion, presumably corresponding to the fully anion exchanged macrocycle. This is indicative of strong binding but precluded the calculation of an association constant. HSO4− on the other hand was bound with modest strength, and ClO4− not at all. The pKa of HClO4 is the same as that of HI (10 pKa units), indicating that the tetrahedral geometry and more diffuse charge disfavour anion binding compared to the spherical halide.
The linear triatomic anions N3− and SCN− are isoelectronic and have similar pKa values in water (4.7 and 4.0, respectively), however, while macrocycle 1 was found to bind azide the most strongly of the monoanions, thiocyanate was not found to associate at all. The crystal structure of the tetra-azide salt shows the anion bridging two imidazolium units via hydrogen bonds (Fig. 7b). This bridging coordination mode optimises the CH⋯N hydrogen bond distances (2.16 Å) compared to a theoretical model in which the azide anion sits lengthwise within the macrocycle (>3 Å). This result highlights the subtle interplay of size and hydrogen bonding ability which makes the macrocycle complementary to N3− but not SCN−.
Carboxylates display a bifurcated anion geometry which could permit a binding mode in which both of the carboxylate oxygen atoms are coordinated by the two hydrogen bond donors at each end of macrocycle 1. 1H NMR titrations revealed that acetate was indeed bound strongly by 1, and benzoate displayed a slightly lower affinity. Some upfield movement of the phenylene proton (Ha) resonances was observed in the benzoate titration, indicative of a degree of charge-assisted π–π stacking. This form of interaction was confirmed by a highly hydrated crystal structure (Fig. 7d) in which some of the benzoate units reside with their phenyl groups inside the macrocycle cavity.
The shape specificity of the system was also probed using the planar chloroaurate and chloropalladate anions. Addition of tetrachloroaurate as its TBA salt to 1 in DMSO-d6 surprisingly produced a small upfield shift (Δδ10eqv = −0.060 ppm) in the Hb resonance, and minor changes in the other proton peaks. This unusual behaviour remained unexplained until crystallographic data were collected (Fig. 7c). X-ray structure analysis revealed that the anion had been reduced to AuCl2−, releasing two additional chloride anions. This was consistent with the observation that while the TBA AuCl4 salt was yellow (Au3+), the crystals were colourless (Au+). The cause of the reduction is unclear, although the most likely candidate is concomitant oxidation of the solvent (DMF or DMSO). The linear AuCl2− unit resides in the very centre of the macrocycle, making short contacts with the phenylene carbons. Precisely whether this should be considered a case of electrostatics working in tandem with packing requirements, an anion–π interaction, or a gold–π interaction54 is unclear. Titrating macrocycle 1 with TBA AuCl2 in DMSO-d6 resulted in an upfield progression of the macrocycle's imidazolium Hb peak (Δδ = −0.174). This peak behaviour is consistent with the AuCl2− anion residing inside the cavity and participating in anion–π interactions as seen in the crystal structure.
A titration was also performed using TBA2 Pd2Cl6 in DMSO-d6. Very strong binding was observed, with the macrocycle proton resonance perturbations being similar to those noted for the halides. No evidence of reduction of the palladate dianion was observed, which is consistent with the greater ‘noble’ character of gold. Strong binding is expected in this case because of the dinegative charge of the palladate anion.
In summary, macrocycle 1 displays a strong affinity for anions, importantly showing no signs of irreversible decomposition, in contrast to CBPQT4+. Selectivity is determined by a combination of geometric and electronic factors. While the acidic imidazolium Hb protons offer hydrogen bonds to more basic anions, anion–π mediated binding is also displayed. Whereas in solution it is likely that anion binding occurs by inclusion, in the solid state the formation of networks prevails. Of the singly charged anions, the strongest association was observed with the linear azide anion.
Conclusions
A highly stable tetra-cationic imidazoliophane 1 has been developed. It has been structurally characterised in depth, revealing its breadth of conformational flexibility. Studies with electron rich neutral guests have elucidated a number of binding modes possible with 1. In particular, complexes are formed with 1,5-dihydroxynaphthalene derivatives, leading to well defined inclusion complexes, demonstrating the potential for use in functional rotaxanes and catenanes analogous to those of CBPQT4+. Anion binding studies show that 1 forms complexes in competitive media, with the binding strength being a function of anion basicity, geometry, and charge. Importantly, anions are bound much more strongly than neutral π-electron rich systems, a feature which opens up the possibility of anion controlled shuttling rotaxanes (currently under investigation).Tetrakis-imidazolium macrocycle 1 is thereby established as an alternative to CBPQT4+, comparing favourably in terms of stability, and breadth of function.55
Acknowledgements
C. J. S. thanks Johnson Matthey and the EPSRC for a CASE studentship. We are grateful to Diamond Light Source for the award of beamtime (MT1880/MT1808). We thank Dr Qiang Zeng for electrochemical analysis.Notes and references
- A. C. Benniston, A. Harriman, D. Philp and J. F. Stoddart, J. Am. Chem. Soc., 1993, 115, 5298–5299 CrossRef CAS.
- W.-Q. Deng, A. H. Flood, J. F. Stoddart and W. A. Goddard, J. Am. Chem. Soc., 2005, 127, 15994–15995 CrossRef CAS.
- E. Cordova, R. A. Bissell, N. Spencer, P. R. Ashton, J. F. Stoddart and A. E. Kaifer, J. Org. Chem., 1993, 58, 6550–6552 CrossRef CAS.
- A. Mirzoian and A. E. Kaifer, J. Org. Chem., 1995, 60, 8093–8095 CrossRef CAS.
- D. Philp, A. M. Z. Slawin, N. Spencer, J. F. Stoddart and D. J. Williams, J. Chem. Soc., Chem. Commun., 1991, 1584–1586 RSC.
- T. T. Goodnow, M. V. Reddington, J. F. Stoddart and A. E. Kaifer, J. Am. Chem. Soc., 1991, 113, 4335–4337 CrossRef CAS.
- A. R. Bernardo, J. F. Stoddart and A. E. Kaifer, J. Am. Chem. Soc., 1992, 114, 10624–10631 CrossRef CAS.
- J. F. Stoddart, Chem. Soc. Rev., 2009, 38, 1802–1820 RSC.
- R. Hernandez, H.-R. Tseng, J. W. Wong, J. F. Stoddart and J. I. Zink, J. Am. Chem. Soc., 2004, 126, 3370–3371 CrossRef CAS.
- A. H. Flood, J. F. Stoddart, D. W. Steuerman and J. R. Heath, Science, 2004, 306, 2055–2056 CrossRef CAS.
- Q. Li, C. H. Sue, S. Basu, A. Shveyd, W. Zhang, G. Barin, L. Fang, A. Sarjeant, J. Stoddart and O. Yaghi, Angew. Chem., Int. Ed., 2010, 49, 6751–6755 CrossRef CAS.
- I. Aprahamian, W. R. Dichtel, T. Ikeda, J. R. Heath and J. F. Stoddart, Org. Lett., 2007, 9, 1287–1290 CrossRef CAS.
- Y.-L. Zhao, A. Trabolsi and J. F. Stoddart, Chem. Commun., 2009, 4844–4846 RSC.
- D. B. Amabilino, P. R. Ashton, M. S. Tolley, J. F. Stoddart and D. J. Williams, Angew. Chem., Int. Ed. Engl., 1993, 32, 1297–1301 CrossRef.
- M. Asakawa, P. R. Ashton, S. Menzer, F. M. Raymo, J. F. Stoddart, A. J. P. White and D. J. Williams, Chem.–Eur. J., 1996, 2, 877–893 CAS.
- P. R. Ashton, A. M. Heiss, D. Pasini, F. M. Raymo, A. N. Shipway, J. F. Stoddart and N. Spencer, Eur. J. Org. Chem., 1999, 1999, 995–1004 CrossRef.
- F. Vögtle, W. M. Müller, U. Müller, M. Bauer and K. Rissanen, Angew. Chem., Int. Ed. Engl., 1993, 32, 1295–1297 CrossRef.
- A. Brown, K. M. Mullen, J. Ryu, M. J. Chmielewski, S. M. Santos, V. Felix, A. L. Thompson, J. E. Warren, S. I. Pascu and P. D. Beer, J. Am. Chem. Soc., 2009, 131, 4937–4952 CrossRef CAS.
- H.-Y. Gong, B. M. Rambo, E. Karnas, V. M. Lynch and J. L. Sessler, Nat. Chem., 2010, 2, 406–409 CrossRef CAS.
- C. Radloff, H.-Y. Gong, C. Schulte to Brinke, T. Pape, V. M. Lynch, J. L. Sessler and F. E. Hahn, Chem.–Eur. J., 2010, 16, 13077–13081 CrossRef CAS.
- B. Odell, M. V. Reddington, A. M. Z. Slawin, N. Spencer, J. F. Stoddart and D. J. Williams, Angew. Chem., Int. Ed. Engl., 1988, 27, 1547–1550 CrossRef.
- C. Henri-Jean, P. C. Pascal, S. Jean-Francis and T. Marc, Chem.–Eur. J., 2004, 10, 5607–5622 CrossRef CAS.
- C.-H. Sue, S. Basu, A. C. Fahrenbach, A. K. Shveyd, S. K. Dey, Y. Y. Botros and J. F. Stoddart, Chem. Sci., 2010, 1, 119–125 RSC.
- A. Altomare, G. Cascarano, C. Giacovazzo, A. Guagliardi, M. C. Burla, G. Polidori and M. Camalli, J. Appl. Crystallogr., 1994, 27, 435 CrossRef.
- P. W. Betteridge, J. R. Carruthers, R. I. Cooper, K. Prout and D. J. Watkin, J. Appl. Crystallogr., 2003, 36, 1487 CrossRef CAS.
- J. Cosier and A. M. Glazer, J. Appl. Crystallogr., 1986, 19, 105–107 CrossRef CAS.
- Z. Otwinowski and W. Minor, Processing of X-ray Diffraction Data Collected in Oscillation Mode, Academic Press, 1997 Search PubMed.
- P. v. d. Sluis and A. L. Spek, Acta Crystallogr., Sect. A: Found. Crystallogr., 1990, A46, 194–201.
- A. Spek, J. Appl. Crystallogr., 2003, 36, 7–13 CrossRef CAS.
- Y.-L. Zhao, L. Liu, W. Zhang, C.-H. Sue, Q. Li, O. S. Miljanicacute, O. M. Yaghi and J. F. Stoddart, Chem.–Eur. J., 2009, 15, 13356–13380.
- M. J. Hynes, J. Chem. Soc., Dalton Trans., 1993, 311–312 RSC.
- D. V. Konarev, V. N. Semkin, A. Graja and R. N. Lyubovskaya, J. Mol. Struct., 1998, 450, 11–22 CrossRef CAS.
- G. Cooke, H. Augier de Cremiers, F. M. A. Duclairoir, M. Gray, P. Vaqueiro, A. V. Powell, G. Rosair and V. M. Rotello, Tetrahedron Lett., 2001, 42, 5089–5091 CrossRef CAS.
- A. Credi, M. Montalti, V. Balzani, S. J. Langford, F. M. Raymo and J. F. Stoddart, New J. Chem., 1998, 22, 1061–1065 RSC.
- P.-L. Anelli, M. Asakawa, P. R. Ashton, R. A. Bissell, G. Clavier, R. Górski, A. E. Kaifer, S. J. Langford, G. Mattersteig, S. Menzer, D. Philp, A. M. Z. Slawin, N. Spencer, J. F. Stoddart, M. S. Tolley and D. J. Williams, Chem.–Eur. J., 1997, 3, 1113–1135 CrossRef CAS.
- D. Curiel, A. Cowley and P. D. Beer, Chem. Commun., 2005, 236–238 RSC.
- M. J. Chmielewski, L. Zhao, A. Brown, D. Curiel, M. R. Sambrook, A. L. Thompson, S. M. Santos, V. Felix, J. J. Davis and P. D. Beer, Chem. Commun., 2008, 3154–3156 RSC.
- L. Zhao, J. J. Davis, K. M. Mullen, M. J. Chmielewski, R. M. J. Jacobs, A. Brown and P. D. Beer, Langmuir, 2009, 25, 2935–2940 CrossRef CAS.
- L. Zhao, K. M. Mullen, M. J. Chmielewski, A. Brown, N. Bampos, P. D. Beer and J. J. Davis, New J. Chem., 2009, 33, 760–768 RSC.
- K. J. Chang, D. Moon, M. S. Lah and K. S. Jeong, Angew. Chem., Int. Ed., 2005, 44, 7926–7929 CrossRef CAS.
- J.-m. Suk and K.-S. Jeong, J. Am. Chem. Soc., 2008, 130, 11868–11869 CrossRef CAS.
- H. Juwarker, J.-m. Suk and K.-S. Jeong, Chem. Soc. Rev., 2009, 38, 3316–3325 RSC.
- M. K. Chae, J.-m. Suk and K.-S. Jeong, Tetrahedron Lett., 2010, 51, 4240–4242 CrossRef CAS.
- H. Greenfield, H. W. Sternberg, R. A. Friedel, J. H. Wotiz, R. Markby and I. Wender, J. Am. Chem. Soc., 1956, 78, 120–124 CrossRef.
- M. A. Neukamm, A. Pinto and N. Metzler-Nolte, Chem. Commun., 2008, 232–234 RSC.
- P. R. Ashton, B. Odell, M. V. Reddington, A. M. Z. Slawin, J. F. Stoddart and D. J. Williams, Angew. Chem., Int. Ed. Engl., 1988, 27, 1550–1553 CrossRef.
- D. Seyferth, T. Kugita, A. L. Rheingold and G. P. A. Yap, Organometallics, 1995, 14, 5362–5366 CrossRef CAS.
- W. W. H. Wong, M. S. Vickers, A. R. Cowley, R. L. Paul and P. D. Beer, Org. Biomol. Chem., 2005, 3, 4201–4208 RSC.
- K. Chellappan, N. J. Singh, I.-C. Hwang, J. W. Lee and K. S. Kim, Angew. Chem., Int. Ed., 2005, 44, 2899–2903 CrossRef CAS.
- C. E. Willans, K. M. Anderson, L. C. Potts and J. W. Steed, Org. Biomol. Chem., 2009, 7, 2756–2760 RSC.
- K. Sato, T. Onitake, S. Arai and T. Yamagishi, Heterocycles, 2003, 60, 779–783 CrossRef CAS.
- S. K. Kim, B.-G. Kang, H. S. Koh, Y. J. Yoon, S. J. Jung, B. Jeong, K.-D. Lee and J. Yoon, Org. Lett., 2004, 6, 4655–4658 CrossRef CAS.
- D. P. Cormode, S. S. Murray, A. R. Cowley and P. D. Beer, Dalton Trans., 2006, 5135–5140 RSC.
- E. R. T. Tiekink and J. Zukerman-Schpector, CrystEngComm, 2009, 11, 1176–1186 RSC.
- Preliminary cyclic voltammetric electrochemical studies have provided further evidence for the stability of macrocycle 1, which remained unreduced in acetonitrile up to the highly reducing potential −2.3 V relative to an Ag/Ag+ electrode.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details and crystal structures in CIF format. CCDC reference numbers 796359–796370, 802075–802076. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c0sc00511h |
| This journal is © The Royal Society of Chemistry 2011 |