Palladium-catalyzed amination reactions in flow: overcoming the challenges of clogging via acoustic irradiation†
Timothy
Noël
a,
John R.
Naber
a,
Ryan L.
Hartman
b,
Jonathan P.
McMullen
b,
Klavs F.
Jensen
*b and
Stephen L.
Buchwald
*a
aDepartment of Chemistry, Massachusetts Institute of Technology, 77 Massachusetts Avenue, Cambridge, Massachusetts 02139, USA. Tel: +1-617-253-1885; Fax: +1-617-253-3297; E-mail: sbuchwal@mit.edu.
bDepartment of Chemical Engineering, Massachusetts Institute of Technology, 77 Massachusetts Avenue, Cambridge, Massachusetts 02139, USA. Tel: +1 617 253 4589; Fax: +1 617 298 8992; E-mail: kfjensen@mit.edu.
First published on 3rd December 2010
Abstract
A continuous-flow palladium-catalyzed amination reaction was made possible through efficient handling of solids via acoustic irradiation. Various diarylamines were obtained with reaction times ranging from 20 s to 10 min.
1. Introduction
The use of microfluidic devices has recently attracted much attention from the pharmaceutical industry.1–3 This is due to the fact that these devices provide several advantages for chemical synthesis, such as enhanced heat- and mass-transfer, safety of operation, the potential for continuous purification, precise control over residence (reaction) time and scalability by operating several devices in parallel (scale-out or numbering-up).4–6Despite these advantages, several challenges still remain. One of the biggest hurdles in the development of flow chemistry methods is the handling of solids. This includes solid reagents,7–12 as well as products and by-products. For example, precipitates can form during the reaction usually leading to irreversible clogging of the microchannels. Several approaches have been suggested to address this problem. The use of segmented liquid–liquid flow can prevent the particles from interacting with the reactor walls.13–16 Although this is an effective way to deal with solids, the use of an additional solvent can reduce the efficiency of the reactions or be incompatible with the reagents. Recently, we have presented a biphasic system of an organic solvent and water, which could solubilize both the organic and inorganic components of a reaction.17,18 However, the presence of water can be detrimental to many reactions. Ultrasonication19,20 and mechanical vibration21 have also been shown to be effective in certain cases.
Palladium-catalyzed C–N cross-coupling reactions constitute a commonly utilized technology in both industry and academia.22–29 This methodology allows the coupling of aryl halides or pseudo halides with a wide variety of nitrogen nucleophiles. These aromatic amines are very common in biologically active molecules30 and, as a result, a method for performing these reactions in flow would be of great interest. However, these reactions form inorganic salts (NaCl, NaBr and NaOTf), which are insoluble in the non-polar solvents typically used for these transformations, including 1,4-dioxane, tetrahydrofuran and di-nbutyl ether. Additionally, when using catalysts based on dialkyl biarylmonophosphines,31–33 these reactions are typically complete in several minutes, resulting in a very fast generation of these insoluble salts. This makes the development of a microfluidic system that can manage these conditions a very challenging task. In this paper, we describe a microfluidic system that is suitable to handle the solid by-products formed in the palladium-catalyzed amination reaction.
Recently, our groups have studied the mechanisms that govern plugging in microfluidic devices.34 Both bridging and constriction were shown to be important mechanisms that lead to clogging. We discovered that bridging could be eliminated via acoustic irradiation while constriction could be managed via fluid velocity. However, to be synthetically viable a more versatile system had to be developed.
2. Results and discussion
A microfluidic system was assembled as shown in Fig. 1. Solutions of the Pd precatalyst 2,35 reagents and NaOtBu in tetrahydrofuran (THF) were loaded into syringes and pumped into the microfluidic system via syringe pumps. It is worth mentioning that initial experiments showed that a steady flow of the NaOtBu base could not be achieved with an HPLC pump, and more reliable flow rates of the reagents were observed with syringe pumps. The precatalyst and reagent streams were combined in a T-mixer prior to the introduction of the NaOtBu stream at a second T-mixer.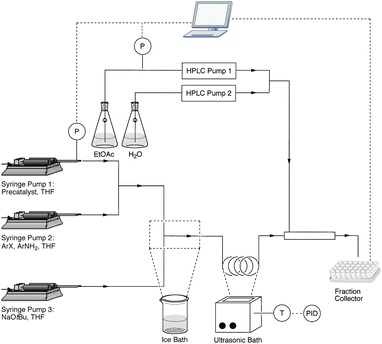 | ||
| Fig. 1 Experimental setup that can handle solids formed during the palladium-catalyzed amination reactions. | ||
This second T-mixer was cooled in an ice bath to prevent any significant reaction from taking place during the mixing process. Without this cooling, the reaction initiated quickly and the salt by-product clogged the T-mixer. Placing the T-mixer in the ultrasonic bath did not lead to any improvement. Next, the reagents were introduced into a 400 μL reactor made of PFA tubing (0.04” inner diameter, 50 cm length). This reactor was placed in an ultrasonic bath heated to 60 °C. Upon exiting the reactor, the reaction was mixed with a quench of water and ethyl acetate (EtOAc) in a piece of larger diameter PFA tubing (2 mm inner diameter, 12.7 cm length, 400 μL volume). This large volume for the quench allowed ample time for the dissolution of the salt by-products. Without this piece of tubing we observed clogging at the end of the reactor. After the quench, the reaction stream was connected to an automated fraction collector. The pumps, both syringe and HPLC, and the fraction collector were controlled via LabVIEW. Software programs were developed to remotely control the pump flow rates and fraction collection and allowed several experiments to be performed in an automated fashion.
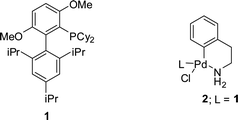
Initially, we conducted the Pd-catalyzed coupling of 4-chloroanisole and aniline with THF as the solvent at 60 °C. The catalytically active L1Pd(0) species was generated starting from the BrettPhos precatalyst 2 (Fig. 2) which is the most active catalyst to date for these type of transformations.31,32 As can be seen from Fig. 3, the reaction was completed in only 60 s with 0.5 mol% of BrettPhos precatalyst. This can be compared to our biphasic method where with 1.2 mol% of BrettPhos precatalyst it took 10 min to obtain full conversion.17 We also compared these results to those obtained in batch experiments. As can be seen from Fig. 3, there was no loss in activity when the reaction was performed in flow. In fact, at very short residence times (e.g., 20 s residence time) we observed higher activity for the reaction performed in flow.36 This result demonstrates one of the major advantages of flow chemistry, and microfluidics specifically, which is the increased heat- and mass-transfer that allows for very fast, efficient heating, which makes it ideally suited for very rapid reactions.
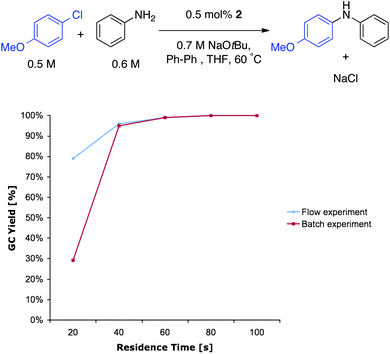 | ||
| Fig. 3 Comparison between flow and batch experiments. | ||
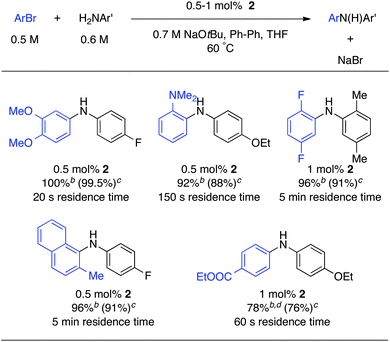
Encouraged by these results, the coupling reactions of several other aryl chlorides were examined with our microfluidic system (Table 1). We were able to couple primary aliphatic amines and aryl chlorides in good yield with a residence time of 5 min with 1 mol% of catalyst. It is noteworthy that these longer residence times were well tolerated by our system. No clogging due to constriction or bridging was observed at this low flow rate. Also ortho substituents were well tolerated: the reaction went to full conversion in only 60 s. 2-Chloropyridines could be coupled with primary aromatic amines in less than 60 s in our system.
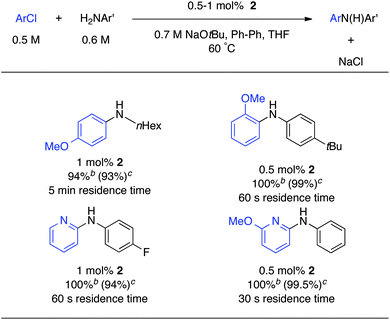
We then turned our attention to aryl bromides (Table 2). These electrophiles have been widely used in the initial studies on Pd-catalyzed aminations because of the ease of the oxidative addition step.37,38 Very good results were obtained in flow for electron-rich substrates: full conversion was obtained in 20 s for non-sterically hindered substrates. Longer residence times were required with substrates bearing ortho substituents, but full conversion was still obtained within 5 min. For aryl bromides bearing esters, weaker bases are typically used in batch processes.39 Although we also observed the formation of a significant amount of the tBu-ester in flow, good yields were obtained for the desired product.
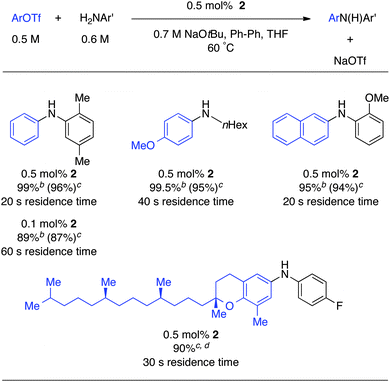
Aryl triflates are generally known to be very reactive substrates in Pd-catalyzed aminations.40 These substrates are attractive since they can be easily synthesized from the corresponding phenols. However, it is known that strong bases such as NaOtBu can mediate triflate hydrolysis leading to lower yields. Therefore weaker bases such as Cs2CO3 are typically used in batch reactions, resulting in slower reactions. With our microfluidic system, we have the advantage that we can control the residence times very accurately and can do both the reaction and the workup in flow in a matter of seconds. Since heat- and mass-transfer are very efficient in microfluidic systems, these reactions can be done very fast before competitive triflate hydrolysis becomes a problem. As can be seen from Table 3, full conversions were obtained in less than 1 min residence time in all cases. Also, biologically active compounds, such as δ-tocopheramines, could be synthesized very efficiently in only 30 s.41,42 Interestingly, when we used a lower catalyst loading, we observed lower yields of the desired product. We hypothesized that lower catalyst loadings lead to slower reaction rates allowing for competitive triflate hydrolysis. These results show that a rigorous control of reaction time and catalyst loading is required to obtain optimal results.
| a ArOTf (0.5 M), ArNH2 (0.6 M), 2 (0.5 mol%), NaOtBu (0.7 M), biphenyl as an internal standard (20 mol%), THF, 60 °C. b GC Yield. c Isolated yield on a 1 mmol scale, reaction went to 100% conversion. d Given the high molecular weight of the corresponding triflate, lower concentrations were required to dissolve the triflate: ArOTf (0.25 M), ArNH2 (0.3 M), 2 (0.5 mol%), NaOtBu (0.35 M), biphenyl as an internal standard (20 mol%), THF, 60 °C. |
|---|
|
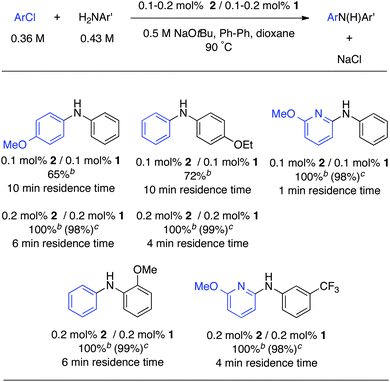
Because we usually obtained full conversion in short reaction times, we investigated lower catalyst loadings in flow. The combination of short reaction times and low catalyst loadings is of great interest to the pharmaceutical industry. To obtain good conversions, the reaction was performed at 90 °C and dioxane was used as a solvent (Table 4). The limited solubility of NaOtBu in dioxane required the use of less concentrated solutions. Nevertheless, with only 0.1 mol% of catalyst loading, we still obtained good conversions with a residence time of 10 min. With a highly reactive substrate, such as 6-chloro-2-methoxypyridine, full conversion could be obtained in only 1 min. Increasing the catalyst loading to 0.2 mol% resulted in a full conversion in several other cases.
Having defined a very robust system that can handle solids in a wide range of conditions, we felt that the next challenge would be conducting a long experiment without clogging. We were able to continuously run the coupling of 4-chloroanisole and aniline in flow with a residence time of 1 min for one hour without any sign of constriction or bridging (Fig. 4).
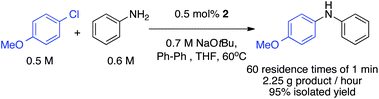 | ||
| Fig. 4 Coupling of 4-chloroanisole with aniline in the presence of 0.5 mol% of catalyst for one hour (1 min residence time). | ||
3. Conclusions
In conclusion, we have developed a fully automated microfluidic system that can efficiently handle solids, even at high concentrations, via acoustic irradiation. We have demonstrated the robustness of this system with the palladium-catalyzed amination reaction of aryl chlorides, aryl bromides and aryl triflates. The amination reaction was followed by an immediate work up in flow. This offered the opportunity to be able to work up reactions quickly and to determine conversions/yields precisely, even at very short reaction times. The latter is something that is difficult to achieve with traditional batch techniques. Finally, we believe that our system provides new opportunities for continuous flow multistep syntheses that require a heterogeneous reaction.Acknowledgements
S.L.B. and K.F.J. thank the Novartis International AG for funding.Notes and references
- G. Davis, Innovations in Pharmaceutical Technology, 2008, 24–27 Search PubMed.
- D. M. Roberge, L. Ducry, N. Bieler, P. Cretton and B. Zimmermann, Chem. Eng. Technol., 2005, 28, 318–323 CrossRef CAS.
- J. Hogan, Nature, 2006, 442, 351–352 CrossRef CAS.
- R. L. Hartman and K. F. Jensen, Lab Chip, 2009, 9, 2495–2507 RSC.
- E. R. Murphy, J. R. Martinelli, N. Zaborenko, S. L. Buchwald and K. F. Jensen, Angew. Chem., Int. Ed., 2007, 46, 1734–1737 CrossRef CAS.
- R. L. Hartman, J. R. Naber, S. L. Buchwald and K. F. Jensen, Angew. Chem., Int. Ed., 2010, 49, 899–903 CAS.
- N. Wang, T. Matsumoto, M. Ueno, H. Miyamura and S. Kobayashi, Angew. Chem., Int. Ed., 2009, 48, 4744–4746 CrossRef CAS.
- J. Kobayashi, Y. Mori, K. Okamoto, R. Akiyama, M. Ueno, T. Kitamori and S. Kobayashi, Science, 2004, 304, 1305–1308 CrossRef CAS.
- S. J. Haswell, B. O'Sullivan and P. Styring, Lab Chip, 2001, 1, 164–166 RSC.
- M. W. Losey, M. A. Schmidt and K. F. Jensen, Ind. Eng. Chem. Res., 2001, 40, 2555–2562 CrossRef CAS.
- M. D. Hopkin, I. R. Baxendale and S. V. Ley, Chem. Commun., 2010, 46, 2450–2452 RSC.
- I. R. Baxendale, J. Deeley, C. M. Griffiths-Jones, S. V. Ley, S. Saaby and G. K. Tranmer, Chem. Commun., 2006, 2566–2568 RSC.
- H. Nagasawa and K. Mae, Ind. Eng. Chem. Res., 2006, 45, 2179–2186 CrossRef CAS.
- H. Song, D. L. Chen and R. F. Ismagilov, Angew. Chem., Int. Ed., 2006, 45, 7336–7356 CrossRef CAS.
- S. L. Poe, M. Q. Cummings, M. P. Haaf and D. T. McQuade, Angew. Chem., Int. Ed., 2006, 45, 1244–1248.
- I. Shestopalov, J. D. Tice and R. F. Ismagilov, Lab Chip, 2004, 4, 316–321 RSC.
- J. R. Naber and S. L. Buchwald, Angew. Chem., Int. Ed., 2010 DOI:10.1002/anie.201004425.
- P. Bazinet, J. P. McMullen, J. R. Naber, A. Musacchio, K. F. Jensen and S. L. Buchwald, Angew. Chem., Int. Ed., 2010 Search PubMed (submitted).
- T. Horie, M. Sumino, T. Tanaka, Y. Matsushita, T. Ichimura and J.-I. Yoshida, Org. Process Res. Dev., 2010, 14, 405–410 Search PubMed.
- J. Sedelmeier, S. V. Ley, I. R. Baxendale and M. Baumann, Org. Lett., 2010, 12, 3618–3621 CrossRef CAS.
- Patent of Japan No. 2006–239502, 2006.
- D. S. Surry and S. L. Buchwald, Chem. Sci., 2011 10.1039/c0sc00331j.
- D. S. Surry and S. L. Buchwald, Angew. Chem., Int. Ed., 2008, 47, 6338–6361 CrossRef CAS.
- N. Marion and S. P. Nolan, Acc. Chem. Res., 2008, 41, 1440–1449 CrossRef CAS.
- J. F. Hartwig, Acc. Chem. Res., 2008, 41, 1534–1544 CrossRef CAS.
- S. Tasler, J. Mies and M. Langa, Adv. Synth. Catal., 2007, 349, 2286–2300 CrossRef CAS.
- C. Torborg and M. Beller, Adv. Synth. Catal., 2009, 351, 3027–3043 CrossRef CAS.
- S. L. Buchwald, C. Mauger, G. Mignani and U. Scholz, Adv. Synth. Catal., 2006, 348, 23–39 CrossRef CAS.
- E. A. B. Kantchev, C. J. O'Brien and M. G. Organ, Angew. Chem., Int. Ed., 2007, 46, 2768–2813 CrossRef CAS.
- D. A. Horton, G. T. Bourne and M. L. Smythe, Chem. Rev., 2003, 103, 893–930 CrossRef CAS.
- B. P. Fors, D. A. Watson, M. R. Biscoe and S. L. Buchwald, J. Am. Chem. Soc., 2008, 130, 13552–13554 CrossRef CAS.
- D. Maiti, B. P. Fors, J. L. Henderson, Y. Nakamura and S. L. Buchwald, Chem. Sci., 2011 10.1039/c0sc00330a.
- X. Huang, K. W. Anderson, D. Zim, L. Jiang, A. Klapars and S. L. Buchwald, J. Am. Chem. Soc., 2003, 125, 6653–6655 CrossRef CAS.
- R. L. Hartman, J. R. Naber, N. Zaborenko, S. L. Buchwald and K. F. Jensen, Org. Process Res. Dev., 2010, 14, 1347–1357 Search PubMed.
- M. R. Biscoe, B. P. Fors and S. L. Buchwald, J. Am. Chem. Soc., 2008, 130, 6686–6687 CrossRef CAS.
- One referee pointed out that the rate-enhancement in the flow reaction could be attributed partly to the acoustic irradiation. Indeed, in the absence of ultrasonication the reaction yield was approximately 6% lower. For a more detailed discussion we refer to ref. 34.
- A. S. Guram, R. A. Rennels and S. L. Buchwald, Angew. Chem., Int. Ed. Engl., 1995, 34, 1348–1350 CrossRef CAS.
- J. Louie and J. F. Hartwig, Tetrahedron Lett., 1995, 36, 3609–3612 CrossRef CAS.
- J. P. Wolfe, H. Tomori, J. P. Sadighi, J. Yin and S. L. Buchwald, J. Org. Chem., 2000, 65, 1158–1174 CrossRef CAS.
- J. P. Wolfe and S. L. Buchwald, J. Org. Chem., 1997, 62, 1264–1267 CrossRef CAS.
- S. Ithoh, S. Nagaoka, K. Mukai, S. Ikesu and Y. Kaneko, Lipids, 1994, 29, 799–802 CrossRef.
- F. Mazzini, T. Netscher and P. Salvadori, Eur. J. Org. Chem., 2009, 2063–2068 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic methods and spectral data. See DOI: 10.1039/c0sc00524j |
| This journal is © The Royal Society of Chemistry 2011 |