Group 9 metal-based inhibitors of β-amyloid (1–40) fibrillation as potential therapeutic agents for Alzheimer's disease†
Bradley Yat-Wah
Man‡
a,
Ho-Man
Chan‡
a,
Chung-Hang
Leung
b,
Daniel Shiu-Hin
Chan
a,
Li-Ping
Bai
b,
Zhi-Hong
Jiang
b,
Hung-Wing
Li
*a and
Dik-Lung
Ma
*a
aDepartment of Chemistry, Hong Kong Baptist University, Kowloon Tong, Hong Kong, China. E-mail: edmondma@hkbu.edu.hk; hwli@hkbu.edu.hk.; Fax: (+852) 3411-7348
bCentre for Cancer and Inflammation Research, School of Chinese Medicine, Hong Kong Baptist University, Kowloon Tong, Hong Kong, China
First published on 25th February 2011
Abstract
We report here the first application of Group 9 metal complexes (i.e.iridium(III) and rhodium(III)) as inhibitors of amyloid fibrillogenesis and as luminescent probes for Aβ1–40peptide. These complexes contained aromatic co-ligands to interact with the hydrophobic residues around the N-terminal domain of the Aβ1–40peptide, as well as solvato co-ligands to allow coordinative bond formation with histidine residues. We demonstrate that these complexes could inhibit Aβ1–40peptide aggregation in vitro, with potency superior to previous metal-based inhibitors reported. Furthermore, we have demonstrated the first example of luminescent detection of Aβ1–40peptides by transition metal complexes.
Introduction
Transition metal complexes have emerged as a viable alternative to organic compounds in pharmaceutical medicine. Platinum, ruthenium or gold metal complexes have been used as therapeutic agents for the treatment of cancer,1 human immunodeficiency virus (HIV)2 and arthritis.3 Metal complexes contain organic ligands bound to the metal center in a precise three-dimensional arrangement. These ligands can be readily modified to tune the selectivity and reactivity of the complex for a particular disease target.4 Furthermore, the preparation of metal complexes is highly modular compared to the often lengthy, linear, and protecting group-laden syntheses of organic molecules. Thus, transition metal complexes can usually be prepared in fewer steps and with greater flexibility for modification during each step of the synthesis. However, while transition metal complexes have been widely utilized for the treatment of cancer, their application to other diseases such as Alzheimer's disease has not been as well explored.Alzheimer's disease (AD) is a common neurodegenerative disease that afflicted 5.3 million people within the United States in 2009, a number which is expected to increase to about 11–16 million people in 2050.5 There is a large body of genetic, pathological and biochemical evidence, which shows that the aggregation of the β-amyloid (Aβ) peptides is linked to neurodegeneration in AD patients.6 One of the therapeutic strategies in the treatment of AD is the development of small molecules, which prevents the misfolding and self-assembling aggregation of monomeric Aβ peptides into the neurotoxic fibrils. Short peptides7 and organic molecules8 have been shown to inhibit the aggregation of Aβ, through the formation of organic or peptide-based aggregates which sequester the Aβ peptides. Nanoparticles, such as copolymeric NiPAM:BAM nanoparticles and functionalized quantum dots have also attracted attention as a potential inhibitors of Aβ1–40 fibrillogenesis as demonstrated by Cabaleiro-Lago et al. and our group previously.9 In the context of transition metal complexes, copper(II), platinum(II) and ruthenium(II) complexes have been shown to bind and inhibit the aggregation of Aβ peptides.10–12 The proposed mechanism for these metal-based inhibitors involves the displacement of the labile co-ligands of the transition metal complex by the imidazoleN-donor of histidine residues, forming a covalent attachment.11 Based on the same principle, a number of research groups has developed metal-based inhibitors for enzymes and histidine rich peptide recognition.13 Furthermore, transition metal complexes often possess interesting photophysical properties, which could allow luminescent visualization of Aβ peptides.
We describe herein the first application of Group 9 (i.e.iridium(III) and rhodium(III)) complexes (Fig. 1a) as inhibitors of β-amyloid (1–40) (Aβ1–40) peptide aggregation. We have previously shown that Group 9 solvato metal complexes such as 1a are able to bind to histidine-rich proteins, and can be applied to luminescent protein staining.14 The solvato complexes 1–3 possess labile co-ligands (i.e.H2O), which can be displaced by the imidazoleN-donor moiety of the histidine amino acid residues of the Aβ1–40peptide (Fig. 1b). Barnham et al. have shown that the presence of aromatic co-ligands on the platinum(II) complex were critical for the inhibition of fibrillogenesis,10 through weak non-covalent interactions with the aromatic side chains around the N-terminal domain of the Aβ1–40peptide. These interactions are recognized as important for the inhibitory activities of both organic and inorganic Aβ1–40inhibitors.8b,10 Thus, we investigated planar aromatic C^N co-ligand such as 2-phenylpyridine (ppy) to enhance selectivity to the N-terminal domain of the Aβ1–40peptide. We envisage that the solvato complexes 1–3 have great potential as luminescent probes for Aβ1–40peptide and as inhibitors against Aβ1–40peptide aggregation.
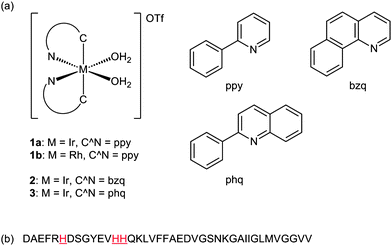 | ||
| Fig. 1 (a) Chemical structures of the solvato complexes 1–3 bearing different metal centers and C^N ligands. (b) Amino acid sequence of the Aβ1–40peptide with the histidine residues highlighted. | ||
Results and discussions
To examine the inhibitory activities of complexes 1–3 against Aβ1–40peptide aggregation, samples of the Aβ1–40peptide monomer were incubated in the presence of different concentrations of the complexes 1–3. Seed-mediated incubation was performed in which small amount of fibrillar Aβ1–40 was sonicated and added into the peptide solution as seed for accelerating the Aβ1–40growth rate.15 After the incubation period, the organic dye Thioflavin T (ThT) was added to label the Aβ1–40fibrils for visualization using highly sensitive total internal reflection fluorescence microscopy (TIRFM) with laser excitation (Fig. 2).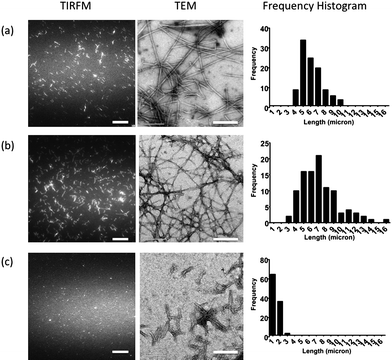 | ||
| Fig. 2 TIRFM fluorescence images (left), TEM images (centre) and the frequency histogram (right) of 50 μM Aβ peptide incubated at 37 °C for 1 h in seed-mediated growth in the presence of: (a) 0 μM; (b) 1 μM; and (c) 5 μM of the complex 1b. The histograms were established by measuring the length of a total of 100 fibrils in each condition. The scale bar for TIRFM and TEM images are 20 μm and 200 nm, respectively. | ||
The fluorescence images of the Aβ1–40peptides incubated in the presence of the iridium (1a, 2 and 3) and rhodium (1b) complexes showed reduced fibril lengths with increasing concentrations of the Group 9 solvato complexes (Fig. 2 and Fig. S1–S3†). These results suggest that the solvato complexes 1–3 could inhibit Aβ1–40peptide aggregation. The inhibitory activity of the complexes 1–3 is presumably due to the coordinative bond formation between the Group 9 metal center and the histidine residues of the Aβ1–40peptide, preventing misfolding of the Aβ1–40peptide and the formation of neurotoxic Aβ1–40fibrils. Of the complexes examined, the rhodium solvato complex 1b showed the highest activity against Aβ1–40peptide aggregation, as shown by the fluorescence and transmission electron microscopy (TEM) images in Fig. 2. Fluorescence and TEM images (Fig. 2) of the fibrils incubated in the presence of 1b exhibited dose-dependent decreases in fibril density and length. When 50 μM Aβ1–40peptides were incubated in the presence of 5 μM of 1b, the resulting images showed fibrils which are significantly reduced in length compared to the controls (Fig. 2a). The median length of the Aβ1–40peptides decreased to approximately 1.3 microns, compared to 6.0 microns for the control sample. The low-density and short fibrils observed in Fig. 2c were expected to be the seeding fibrils added in the beginning of the incubation. The frequency distribution histogram of the length of the Aβ1–40peptide in the presence of 5 μM of 1b is comparable to that of the seed-only conditions (Fig. S5a†). This suggests that nearly complete inhibition of amyloid fibrillogenesis was exhibited at 5 μM of 1b (Fig. 2c). By comparison, complexes 1a, 2 and 3 displayed observable inhibition against fibrillogenesis of Aβ1–40peptides at concentrations of 5–10 μM, >50 μM and 10–17 μM, respectively (Fig. S1–S3†). Interestingly, the solvato complexes bearing C^N co-ligands with extended aromatic systems (i.e.benzoquinoline, 2; phenylquinoline, 3) are less active as inhibitors against fibrillogenesis compared to complex 1a that contain the smaller phenylpyridine aromatic system. One possible explanation for the reduced activities of complexes 2 and 3 could be the steric hindrance caused by the large planar aromatic C^N co-ligands, which prevents the coordination of the N-donor histidine residues with the metal center. Furthermore, we observed that the rhodium(III) complex 1b was more active against the Aβ1–40peptide aggregation than the corresponding isoelectronic iridium(III) complex 1a. To determine the contribution of the metal center and aromatic ligand to the inhibition properties of the complex 1b, Aβ1–40peptides were incubated in the presence of rhodium(III) chloride and the ligand 2-phenylpyridine (ppy) respectively. The resulting TIRFM images showed no significant changes in the median length of the Aβ1–40peptides compared to the control in both cases (Fig. S4†). These results taken together show the importance of binding the planar aromatic ligand to the metal center in the inhibition properties of the group 9 solvato complexes 1–3. Taken together, these results show that the most active compound, the rhodium(III) solvato complex 1b, was able to nearly completely inhibit Aβ1–40peptide fibrillogenesis at 5 μM, with a complex to monomer (c/m) ratio of approximately 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10.
:10.
The low c/m ratio observed in the inhibition experiments suggests that the solvato complexes 1–3 forms a coordinative bond with the seed fibrils and blocks the active elongation site. As a consequence of the coordinative bond formation between the complex and the seed fibrils, an apparently reduced rate of fibrillogenesis was observed. This result is a significant development on the transition metal-based Aβ peptide inhibitors published in literature. The previously reported transition metal-based Aβ inhibitors were mostly designed for Aβ1–42 while that for Aβ1–40 was limited. For example, the platinum(II) complexes developed by Barnham et al. required a c/m ratio of 1:1 for the inhibition of 10 μM of Aβ1–42peptides.10 In addition, the binuclear ruthenium(II)–platinum(II) complex reported by Kumar et al. inhibited 50 μM Aβ1–42 permanently with a c/m ratio around 1:2.16 However, due to the different rates of fibrillogenesis between Aβ1–40 and Aβ1–42,17 the inhibition conditions may not be applicable to both. Our novel application of group 9 solvato metal complexes hereby brings new insight in the development of potential Aβ1–40inhibitor.
The inhibition of Aβ1–40peptides with the metal complexes was found to be instantaneous. In a control experiment, Aβ1–40 monomers were initially incubated for 15 min and yielded short fibrils with median length of ca. 2.5 μm (Fig. S5b). An inhibitive dose (5 μM) of complex 1b was subsequently added to the preformed Aβ1–40fibrils and allowed to incubate for an additional 45 min (i.e. a total incubation time of 1 h). The median length of fibrils at t = 15 min and t = 60 min was measured to be comparable (Fig. S5c†), supporting that the inhibition was readily effective at any instance of complex addition.
To examine the binding of the solvato metal complexes to peptide, ESI-TOF mass spectrometry experiments were performed. Mass spectrometry has been routinely used in the literature to determine the binding mode and stoichiometry of organic and inorganic inhibitors of Aβ1–40peptide aggregation.12,13,18 The mass spectrum of Aβ1–40peptide in the absence of the metal complex (Fig. 3a) exhibited three peaks at 866.8226, 1083.2797 and 1444.0346, corresponding to the 5+, 4+ and 3+ ionization states of the Aβ1–40 monomer, respectively. However, after treatment of the Aβ peptide with the rhodium(III) solvato complex 1b, extra peaks were observed at 948.8379, 1186.0485 and 1580.7290 in the mass spectrum (Fig. 3b), which corresponded to the 1:1 Rh(III)–Aβ1–40 monomer complexes. This data suggests that the rhodium solvato complex 1b is covalently bound to the Aβ1–40peptide, presumably through coordination of the N-donor histidine residue to the rhodium(III) center.
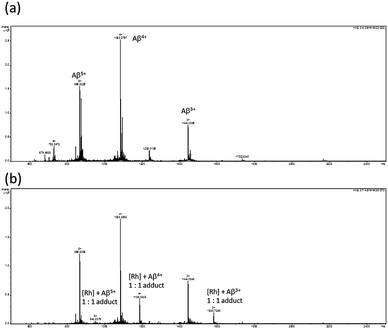 | ||
| Fig. 3 ESI-TOF mass spectra of: (a) Aβ1−40 peptide; and (b) Aβ1−40 peptide incubated in the presence of the rhodium complex 1b. | ||
The cytotoxicity of the most potent complex 1b was examined using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay. Neuroblastoma cells (SH-SY5Y) were incubated in the presence of different concentrations of complex 1b for 24 h, and the cell viability was examined using MTT (Fig. S6†). The IC50 value for the complex 1b was estimated to be between 25–50 μM. The estimated IC50 value against the SH-SY5Y cell line is higher compared to the concentration required for complete inhibition of Aβ1–40peptide aggregation. This suggests the presence of a therapeutic window whereby Aβ1–40 fibrillogenesis can be controlled without significant damage to brain cells.
The iridium(III) solvato complex 1a has been previously demonstrated to function as a luminescent probe for histidine and histidine-rich proteins.14 Thus, we were interested to investigate the effect of varying the extent of conjugation of the C^N co-ligands on the photophysical properties of this type of complex. Due to the auto-fluorescence of the cellular material, an ideal cell-based luminescent probe should emit in the red region of the visible spectrum. All the complexes 1a, 2 and 3 were weakly emissive in aqueous buffer (Fig. 4a), but exhibited a significant enhancement in the emission intensity in the presence of histidine (Fig. 4b). The complex 1b did not show any emission enhancement in the presence of histidine when excited.
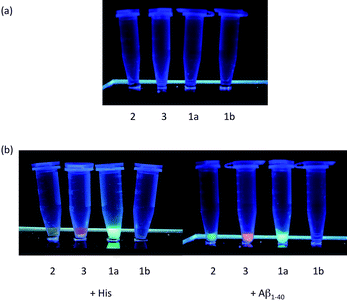 | ||
| Fig. 4 (a) Photographs of complexes 1–3 (25 μM); and (b) complexes 1–3 (25 μM) in the presence of histidine (2.5 mM) and Aβ1−40 fibrils (25 μM, diluted according to monomeric concentration) illuminated using a UV-C lamp respectively. Significant enhancement in fluorescence emission was observed in the presence of histidine and histidine-bearing Aβ1−40 fibrils. | ||
The coordinative bond formation between the solvato complex 1a and histidine-rich peptides shelters the metal center in a hydrophobic environment, reducing solvent-mediated non-radiative decay of the excited state and thereby enhancing phosphorescence. As the aggregated Aβ1–40peptide would be expected to provide a more hydrophobic environment compared to the Aβ1–40 monomer, the emission enhancement of the iridium(III) complexes could be potentially higher when bound to the aggregated form. We observed that in the presence of the Aβ1–40 monomer (Fig. 5b), a fold change of 56 was observed at λmax = 491 nm. By contrast, a fold change of 134 was observed at λmax = 491 nm in the presence of a comparable mass concentration of Aβ1–40fibrils (Fig. 5c). To our knowledge, this is the first example of a transition metal complex that displays a ‘switch-on’ luminescence response upon binding to Aβ1–40peptides. Emission spectra of the complex 1b in the presence of Aβ1–40 monomer and fibrils were also measured, where no emission enhancements were observed (not shown). The magnitude of the ‘switch-on’ luminescence response can be used to differentiate between the monomeric and fibrillar forms of the Aβ1–40peptide.
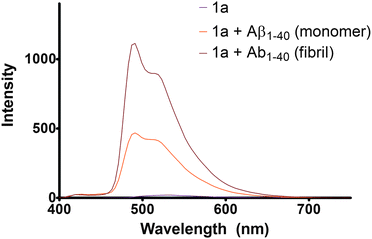 | ||
Fig. 5
Emission spectra of (a) 25 μM of complex 1a ( ); (b) 25 μM Aβ1–40peptide monomer in 25 μM of complex 1a ( ); (b) 25 μM Aβ1–40peptide monomer in 25 μM of complex 1a ( ) and (c) 1 h incubated Aβ1–40fibrils (with 25 μM in monomeric concentration) in 25 μM complex 1a ( ) and (c) 1 h incubated Aβ1–40fibrils (with 25 μM in monomeric concentration) in 25 μM complex 1a ( ). Excitation wavelength = 365 nm. ). Excitation wavelength = 365 nm. | ||
Conclusion
In conclusion, we have successfully synthesized a series of novel cyclometallated iridium(III) and rhodium(III) solvato complexes with different C^N co-ligands. These novel iridium(III) and rhodium(III) complexes were found to inhibit the aggregation of Aβ1–40peptide as revealed by fluorescence and transmission electron microscopy. The complexes (1) containing the phenylpyridine co-ligand were found to be more active than complexes bearing the bulkier benzoquinoline (2) or phenylquinoline (3) co-ligands, presumably due to steric effects. The rhodium(III) solvato complex 1b was the most active complex of the series and was found to nearly completely inhibit Aβ1–40peptide aggregation at 5 μM, an improvement compared to inorganic inhibitors reported in the literature. Using ESI-TOF mass spectrometry, we showed the rhodium complex 1b was bound to the Aβ1–40peptide through covalent interactions, presumably via coordination of the N-donor functionality of the histidine residues. We have also demonstrated that the iridium(III) complexes 1a, 2 and 3 are able to function as luminescent probes for Aβ1–40peptides. Complexes 2 and 3 containing extended aromatic co-ligands exhibited an interesting red-shift in their emission maxima, making them ideal for cellular labeling applications. Furthermore, these solvato complexes could distinguish between the different forms of Aβ1–40peptide, with the Aβ1–40fibrils giving a higher emission enhancement compared to a similar mass concentration of the Aβ1–40 monomers. To our knowledge, this is the first application of a transition metal complex for luminescent sensing of Aβ1–40 aggregates. Thus, the Group 9 complexes presented in this work have great potential as inhibitors of Aβ1–40peptide aggregation, and as labels for the Aβ1–40peptide.This work was supported by a General Research Fund from the Research Grant Councils of the Hong Kong Administrative Region (HKBU201208) and the Faculty Research Grant of the Hong Kong Baptist University (FRG2/09-10/070).
Notes and references
- (a) T. W. Hambley, Dalton Trans., 2007, 4929–4937 RSC; (b) L. Ronconi and P. J. Sadler, Coord. Chem. Rev., 2007, 251, 1633–1648 CrossRef CAS; (c) P. C. A. Bruijnincx and P. J. Sadler, Curr. Opin. Chem. Biol., 2008, 12, 197–206 CrossRef CAS; (d) P. C. A. Bruijnincx and P. J. Sadler, Adv. Inorg. Chem., 2009, 61, 1–62 CAS; (e) A. V. Klein and T. W. Hambley, Chem. Rev., 2009, 109, 4911–4920 CrossRef CAS; (f) K. S. Lovejoy and S. J. Lippard, Dalton Trans., 2009, 10651–10659 RSC; (g) D.-L. Ma, C.-M. Che and S.-C. Yan, J. Am. Chem. Soc., 2009, 131, 1835–1846 CrossRef CAS; (h) P. Wang, C.-H. Leung, D.-L. Ma, S.-C. Yan and C.-M. Che, Chem.–Eur. J., 2010, 16, 6900–6911 CrossRef CAS.
- A. N. Vzorov, D. Bhattacharyya, L. G. Marzilli and R. W. Compans, Antiviral Res., 2005, 65, 57–67 CrossRef CAS.
- (a) M. J. Abrams and B. A. Murrer, Science, 1993, 261, 725–730 CrossRef CAS; (b) L. Messori and G. Marcon, Met. Ions Biol. Syst., 2004, 41, 279–304 CAS; (c) E. Viry, E. Battaglia, V. Deborde, T. Muller, R. Reau, E. Davioud-Charvet and D. Bagrel, ChemMedChem, 2008, 3, 1667–1670 CrossRef CAS.
- (a) S. P. Fricker, Dalton Trans., 2007, 4903–4917 RSC; (b) S. P. Fricker, Metallomics, 2010, 2, 366–377 RSC; (c) Savvas N. Georgiades, Nurul H. Abd Karim, K. Suntharalingam and R. Vilar, Angew. Chem., Int. Ed., 2010, 49, 4020–4034 CAS.
- M. Citron, Nat. Rev. Drug Discovery, 2010, 9, 387–398 CrossRef CAS.
- (a) J. Hardy and D. J. Selkoe, Science, 2002, 297, 353–356 CrossRef CAS; (b) D. J. Selkoe and D. Schenk, Annu. Rev. Pharmacol. Toxicol., 2003, 43, 545–584 CrossRef CAS; (c) D. M. Walsh and D. J. Selkoe, J. Neurochem., 2007, 101, 1172–1184 CrossRef CAS.
- (a) G. Yamin, P. Ruchala and D. B. Teplow, Biochemistry, 2009, 48, 11329–11331 CrossRef CAS; (b) M. Taylor, S. Moore, J. Mayes, E. Parkin, M. Beeg, M. Canovi, M. Gobbi, D. M. A. Mann and D. Allsop, Biochemistry, 2010, 49, 3261–3272 CrossRef CAS.
- (a) T. Cohen, A. Frydman-Marom, M. Rechter and E. Gazit, Biochemistry, 2006, 45, 4727–4735 CrossRef CAS; (b) A. A. Reinke and J. E. Gestwicki, Chem. Biol. Drug Des., 2007, 70, 206–215 CrossRef CAS; (c) H.-S. Hong, S. Rana, L. Barrigan, A. Shi, Y. Zhang, F. Zhou, L.-W. Jin and D. H. Hua, J. Neurochem., 2009, 108, 1097–1108 CrossRef CAS; (d) A. A. Reinke, P. M. U. Ung, J. J. Quintero, H. A. Carlson and J. E. Gestwicki, J. Am. Chem. Soc., 2010, 132, 17655–17657 CrossRef CAS.
- (a) C. Cabaleiro-Lago, F. Quinlan-Pluck, I. Lynch, S. Lindman, A. M. Minogue, E. Thulin, D. M. Walsh, K. A. Dawson and S. Linse, J. Am. Chem. Soc., 2008, 130, 15437–15443 CrossRef CAS; (b) L. Xiao, D. Zhao, W.-H. Chan, M. M. F. Choi and H.-W. Li, Biomaterials, 2009, 31, 91–98.
- S. Lim, B. M. Paterson, M. T. Fodero-Tavoletti, G. J. O'Keefe, R. Cappai, K. J. Barnham, V. L. Villemagne and P. S. Donnelly, Chem. Commun., 2010, 46, 5437–5439 RSC.
- K. J. Barnham, V. B. Kenche, G. D. Ciccotosto, D. P. Smith, D. J. Tew, X. Liu, K. Perez, G. A. Cranston, T. J. Johanssen, I. Volitakis, A. I. Bush, C. L. Masters, A. R. White, J. P. Smith, R. A. Cherny and R. Cappai, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 6813–6818 CrossRef CAS.
- D. Valensin, P. Anzini, E. Gaggelli, N. Gaggelli, G. Tamasi, R. Cini, C. Gabbiani, E. Michelucci, L. Messori, H. Kozlowski and G. Valensin, Inorg. Chem., 2010, 49, 4720–4722 CrossRef CAS.
- (a) A. Y. Louie and T. J. Meade, Chem. Rev., 1999, 99, 2711–2734 CrossRef CAS; (b) S. Sun, F. M. Abul, B. C. Roy and S. Mallik, Org. Lett., 2000, 2, 911–914 CrossRef CAS; (c) A. S. Harney, J. Lee, L. M. Manus, P. Wang, D. M. Ballweg, B. C. La and T. J. Meade, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 13667–13672 CrossRef CAS; (d) K. L. Haas and K. J. Franz, Chem. Rev., 2009, 109, 4921–4960 CrossRef CAS.
- D.-L. Ma, W.-L. Wong, W.-H. Chung, F.-Y. Chan, P.-K. So, T.-S. Lai, Z.-Y. Zhou, Y.-C. Leung and K.-Y. Wong, Angew. Chem., Int. Ed., 2008, 47, 3735–3739 CrossRef CAS.
- T. Ban, D. Hamada, K. Hasegawa, H. Naiki and Y. Goto, J. Biol. Chem., 2003, 278, 16462–16465 CrossRef CAS.
- A. Kumar, L. Moody, J. F. Olaivar, N. A. Lewis, R. L. Khade, A. A. Holder, Y. Zhang and V. Rangachari, ACS Chem. Neurosci., 2010, 1, 691–701 Search PubMed.
- S. W. Snyder, U. S. Ladror, W. S. Wade, G. T. Wang, L. W. Barrett, E. D. Matayoshi, H. J. Huffaker, G. A. Krafft and T. F. Holzman, Biophys. J., 1994, 67, 1216–1228 CrossRef CAS.
- F. N. Bazoti, A. Tsarbopoulos, K. E. Markides and J. Bergquist, J. Mass Spectrom., 2005, 40, 182–192 CrossRef CAS.
Footnotes |
| † Electronic Supplementary Information (ESI) available: Experimental procedures, NMR characterization, fluorescence images, frequency histograms and MTT assay results. See DOI: 10.1039/c0sc00636j/ |
| ‡ B. Y.-W. M. and H.-M. C. contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2011 |