Single-molecule, single-particle observation of size-dependent photocatalytic activity in Au/TiO2 nanocomposites†
Nan
Wang
ab,
Takashi
Tachikawa
*a and
Tetsuro
Majima
*a
aThe Institute of Scientific and Industrial Research (SANKEN), Osaka University, Mihogaoka 8-1, Ibaraki, Osaka 567-0047, Japan. E-mail: tachi45@sanken.osaka-u.ac.jp; majima@sanken.osaka-u.ac.jp; Fax: +81-6-6879-8499; Tel: +81-6-6879-8495
bCollege of Chemistry and Chemical Engineering, Huazhong University of Science and Technology, Wuhan, 430074, P.R. China
First published on 4th March 2011
Abstract
Electronic communication between the building blocks of nanocomposites is an important property that affects their functionality with regard to many optoelectronic and catalytic applications. Herein, we report a single-molecule, single-particle approach for elucidating the inherent photocatalytic activity of individual Au nanoparticle-loaded TiO2 particles using a novel redox-responsive fluorescent dye. A single-particle kinetic analysis of the fluorescence bursts emitted from the products revealed that the photocatalytic activity leading to reduction of the probe molecules is controlled by not only the substrate concentration and excitation intensity but also the Au particle size, and that these factors are intricately interrelated. Furthermore, we discovered that the stochastic photocatalytic events around the millisecond-to-second time scale showed considerable temporal and spatial heterogeneity during photoirradiation, and that they actually originate from the charging/discharging of Au nanoparticles on TiO2. Our findings represent a significant contribution to the scientific understanding of the interfacial electron transfer dynamics in composite systems, and more fundamentally, in heterogeneous (photo)chemical processes.
Introduction
Interfacial electron transfer (ET) is a fundamental process in physics, chemistry, and biology, playing a crucial role in natural and artificial energy conversion systems.1 For instance, charge transport across the heterogeneous interface of metal–semiconductor and semiconductor–semiconductor nanocomposites has attracted a great deal of research interest, because this process largely governs the performance of photovoltaic devices, batteries, fuel cells, sensors, and (photo)catalysts used for water splitting, organic synthesis, and environmental purification.2–6 To facilitate ET reactions, possible rate-limiting factors must be addressed and suitably optimized. The incorporation of noble metals with a large work function (e.g., Au and Ag) onto metal oxide supports (e.g., TiO2 and ZnO) has been shown to significantly enhance the charge separation efficiency via ET from the conduction band (CB) of semiconductor metal oxides to metals on the surface. This results in the Fermi energy level of the loaded metal in the photostationary state (EF*) increasing and eventually reaching equilibrium with that of the metal oxides being subjected to photoirradiation.7 This concept was further developed by Kamat et al., who found that the EF* increased with a decrease in the diameter of the Au nanoparticles (dAu) added to the TiO2 nanoparticles using 3-mercaptopropionic acid as the bifunctional linker. They attributed this observation to the quantum size effect of the Au nanoparticles (3 < dAu < 8 nm).8,9 In addition, Tada et al. reported a converse trend in which the EF* of Au nanoparticles directly deposited on TiO2 increased with dAu in the range of 3 < dAu < 13 nm. They concluded that the predominant mechanisms involved in determining EF* are either the solvation of charged metal nanoparticles in polar media or the quantum size effect in less polar media.10Although a number of studies have been conducted to elucidate the mechanism of photoinduced ET reactions in these composite systems, the directional flow of photogenerated charge carriers through the interface has still not been fully explained because of the inherent static and dynamic heterogeneities in these systems. In fact, recent spectroscopic studies conducted using advanced microscopic techniques have demonstrated that chemical reactions occurring at the surface of a solid are intrinsically heterogeneous and closely related to the structural dispersions and spatial distributions of reactive sites, as well as surface restructuring dynamics, conformations of adsorbates, and electronic interactions between adjacent components.11–22 However, to the best of our knowledge, no single-molecule studies have described the interfacial ET dynamics in nanocomposite systems.
In this study, we investigated the photocatalytic redox reaction over Au nanoparticle-loaded TiO2 (Au/TiO2) particles at the single-molecule, single-particle levels using a novel redox-responsive fluorescent dye. This investigation revealed the temporal and spatial heterogeneities of the interfacial ET process on an individual particle. The single-particle kinetic analysis of the fluorescence characteristics of the product revealed that the substrate concentration, UV light intensity, and structural characteristics of Au nanoparticles are important parameters governing the photocatalytic activity of Au/TiO2. Furthermore, we discovered that the stochastic photocatalytic events actually originate from the charging/discharging of Au nanoparticles on TiO2. Overall, this study provides a great deal of valuable information that is impossible to obtain from ensemble-averaged experiments because each particle behaves differently.
Results and discussion
Bulk photocatalytic experiments
The fluorogenic reaction is based on our recently developed ET-involved reduction of boron-dipyrromethene compound, 3,4-dinitrophenyl-BODIPY (DN-BODIPY, Fig. 1).21 The reduction of DN-BODIPY proceeds along the following general pathway: (i) the nitro (NO2) group at the para-position is first reduced by two electrons to form a nitroso (NO) group, (ii) the NO group immediately accepts another two electrons to form a hydroxylamino (NHOH) group, and (iii) the NHOH group finally becomes an amino (NH2) groupvia a slow two-electron reduction.23,24 Because the two NO2 groups greatly reduce the lowest unoccupied molecular orbital (LUMO) energy level of the benzene moiety introduced at the meso-position of the BODIPY core, the BODIPY fluorescence is significantly quenched by an intramolecular ET from the excited fluorophore to the nitro-substituted benzene moiety prior to reduction.25,26 However, after one of the NO2 groups of DN-BODIPY is reduced preferentially, the other NO2 group neutralizes the increased electron density owing to the produced NHOH or NH2 group, which dramatically suppresses the intramolecular ET process. Therefore, by accepting electrons, non-fluorescent DN-BODIPY (Φfl ≈ 10−4 in methanol) can be reduced to form the highly fluorescent 4-hydroxyamino-3-nitrophenyl-BODIPY (HN-BODIPY, Φfl = 0.50 in methanol), which provides the basis for studying the interfacial ET process on individual nanoparticlesviadetection of the fluorescence of the produced HN-BODIPY at the single-molecule level.21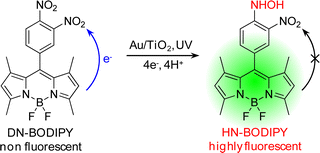 | ||
| Fig. 1 Photocatalytic generation of fluorescent HN-BODIPY from non-fluorescent DN-BODIPY. | ||
The photocatalytic activity of Au/TiO2 particles was first evaluated by ensemble-averaged spectroscopy. We selected three types of Au/TiO2 photocatalysts with the same crystalline anatase form of TiO2 and the same loading amount of Au, but different dAu: 5 nm, 8 nm, and 14 nm Au/TiO2 (Fig. S1†). The structural and optical properties of the selected compounds are listed in Table 1. As the Au particle size increased, the mean number of Au nanoparticles loaded per TiO2 particle (NAu) decreased significantly. As shown in Fig. S2†, Au/TiO2 reduced DN-BODIPY to HN-BODIPYvia the reduction pathway in Ar-saturated methanol under UV light irradiation, where the photogenerated valence band holes (h+) in TiO2 are efficiently scavenged by methanol.27,28 These findings were similar to those observed for bare TiO2 particles,21 but all of the tested Au/TiO2 samples showed much higher photocatalytic reduction ability than TiO2. This difference was likely because the loaded Au nanoparticles greatly enhance the charge separation within the nanocomposites by collecting electrons from TiO2.
| Sample | Annealing conditions | Properties of Au/TiO2 | K ad / μM | γ eff / s −1 | n c | |||
|---|---|---|---|---|---|---|---|---|
| T/°C | t/h | λ max/nm | d Au /nm (RSD, %) | N Au a | ||||
| a N Au represents the mean number of Au nanoparticles loaded per TiO2 particle. b Obtained from Fig. 5D. γeff represents the combined reactivity of all surface catalytic sites. c n is the slope of the log(<τoff>s−1)–log(IUV) plot obtained from Fig. 6A. | ||||||||
| TiO2 | N/A | N/A | N/A | N/A | N/A | 0.5 ± 0.1 | 0.55 ± 0.07 | 0.22 |
| 5 nm Au/TiO2 | 300 | 0.5 | 530 | 5.5 ± 1.2 (21.8) | 21.9 ± 5.7 | 1.1 ± 0.2 | 0.61 ± 0.13 | 0.06 |
| 8 nm Au/TiO2 | 600 | 4.0 | 544 | 8.1 ± 1.5 (18.5) | 10.4 ± 2.8 | 1.0 ± 0.2 | 0.57 ± 0.11 | 0.11 |
| 14 nm Au/TiO2 | 700 | 4.5 | 572 | 14.1 ± 3.2 (22.7) | 3.0 ± 1.5 | 0.8 ± 0.2 | 0.62 ± 0.15 | 0.24 |
Single-particle observation of photocatalytic reduction
Total internal reflection fluorescence microscopy (TIRFM) was used to investigate the ET-induced photocatalytic reduction of DN-BODIPY over individual Au/TiO2 particles. To obtain individual Au/TiO2 nanoparticles, well-dispersed methanol suspensions of Au/TiO2 with very low concentrations were spin-coated on the cleaned cover glasses, which resulted in only one particle being present in a 12.5 μm × 12.5 μm area. Here, the term “single (or individual) Au/TiO2 (nano)particle” indicates one Au/TiO2 particle integrated with several Au nanoparticles present on the surface of one TiO2 nanoparticle.Fig. 2A shows typical fluorescence images captured for a single 8 nm Au/TiO2 particle in Ar-saturated methanol containing DN-BODIPY (2.0 μM) under 488 nm laser and UV irradiation. Individual particles showed a number of fluorescence bursts that had signals higher than the background (also see Fig. 5A). Control experiments confirmed that Au/TiO2 (or TiO2), DN-BODIPY, and UV excitation are essential to the generation of fluorescence bursts. The locations of the fluorescence bursts, which were determined by fitting two-dimensional Gaussian functions to the distribution of fluorescent spots, are likely distributed over the particle (see the red dots in the transmission image of Fig. 2A). Moreover, the fluorescence lifetimes of the in situ generated bursts over individual Au/TiO2 particles were measured by combining confocal microscopy with a time-correlated single-photon counting (TCSPC) system. The fluorescence decay profiles were well-fitted by the biexponential function: a1·exp(τ/τ1) + a2·exp(τ/τ2). As shown in Fig. 2C, the fluorescence burst (component 2) depicted in red in Fig. 2B exhibited a much longer lifetime than the background signal (component 1) from DN-BODIPY in solution (see blue mark in Fig. 2B). These findings suggest that such a sudden increase in intensity corresponds to the generation of the fluorescent product (i.e., HN-BODIPY). However, when compared to free HN-BODIPY molecules in the bulk solution (τ2 = 3.7 ns), these in situ-generated products on the Au/TiO2 surface showed much shorter lifetimes (τ2 = 1–2 ns). This difference may have been due to the intermolecular ET from the excited BODIPY chromophore to the TiO2 and/or Au nanoparticles.21 Although the fluorescence quenching and/or enhancement of HN-BODIPY by Au nanoparticles might be considered, detailed studies to clarify the underlying mechanisms are beyond the scope of this article.
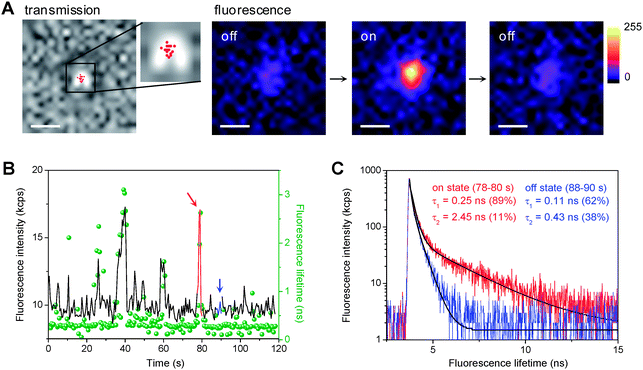 | ||
| Fig. 2 (A) Optical transmission of a single 8 nm Au/TiO2 particle immobilized on a cover glass and fluorescence images of the same particle in Ar-saturated DN-BODIPY solution (2.0 μM in methanol) under 488 nm laser (I488 = 0.1 kW cm−2) and UV irradiation (IUV = 0.5 W cm−2). The fluorescence images display a series of successive images exhibiting on and off events. The acquisition time of an image was 50 ms. The scale bars are 500 nm. The red dots in the transmission image indicate the location of fluorescence bursts. The accuracy of the location was about 20–30 nm. (B) Typical trajectories of fluorescence intensity (black) and lifetime (green, bin time is 500 ms) observed for a single 8 nm Au/TiO2 particle in Ar-saturated DN-BODIPY solution (2.0 μM, methanol) under 485 nm laser (I488 = 2 kW cm−2) and UV irradiation (IUV = 0.5 W cm−2). (C) Fluorescence decay profiles of the burst emission and background emission in the time regions indicated by red and blue in panel B, respectively. Black lines indicate biexponential curves fitted to the data. | ||
Conversely, each decrease in intensity marks the disappearance of the fluorescent product from the surface of the nanoparticle. Our previous experiments showed that photobleaching or blinking of single HN-BODIPY molecules occurs on much longer time scales under similar laser intensities,21 which suggests that the observed sudden decreases in intensity are attributed to either the consecutive reduction or the dissociation of adsorbed HN-BODIPY. Although the occurrence of the fluorescence bursts generated from one Au/TiO2 nanoparticle is stochastic in nature, their statistical properties should provide valuable information regarding the kinetic mechanism of photocatalysis over Au/TiO2 at the single-particle level.
Temporal and spatial heterogeneities of photocatalytic activity
Fig. 3A shows the time profile for the turnover rate of the on-off cycle every 10 s (TOF) over one Au/TiO2 particle during 10 min of UV irradiation (see Section S4 for the method used to calculate TOF†). It is clear that the TOF values over the same particle vary temporally, which is indicative of the fluctuation in the photocatalytic activity of one particle at different times. To quantify the fluctuation in activity, the mean value of the turnover rate (<TOF>) and its relative standard derivation (RSD) over each particle were calculated as shown in Fig. 3B. Based on the statistical analysis of 10 different particles, most of the bare TiO2 particles showed lower activity (<TOF>, 0.23–0.60 s−1) and greater temporal variation (RSD, 30%–51%) in the photoreduction of DN-BODIPY than Au/TiO2 samples. Moreover, bare TiO2 particles possess a much broader distribution in activity. Indeed, approximately 20% of the TiO2 particles did not show any detectable photocatalytic activity (for more than 20 individual particles examined), while approximately 10% of the TiO2 particles showed excellent activity (e.g., see Fig. 3B). These results suggest that there was heterogeneous activity among particles, which is always masked in the ensemble-averaged measurements. Conversely, almost all of the Au/TiO2 particles could effectively photocatalyze the reduction of DN-BODIPY, implying that Au nanoparticles can preferentially trap electrons from TiO2 and then deliver them to DN-BODIPY. In other words, the reduction of DN-BODIPY occurs more easily on the loaded Au nanoparticles. The surface of TiO2 has a number of electron trapping sites with different energies and structures, which results in an extremely broad distribution in the reaction rates;22,29–31 therefore, three different sized Au/TiO2 samples showed smaller fluctuations in activity than bare TiO2 particles (Fig. 3B). Consequently, the precise positions of fluorescent spots that appear on the surface of single particles were analyzed.![(A) Fluctuation of TOF over a single 5 nm Au/TiO2 particle ([DN-BODIPY] = 2.0 μM, IUV = 0.5 W cm−2). (B) Distribution of <TOF> and its RSD over 10 different individual particles for TiO2 (black), 5 nm Au/TiO2 (blue), 8 nm Au/TiO2 (green), and 14 nm Au/TiO2 (red).](/image/article/2011/SC/c0sc00648c/c0sc00648c-f3.gif) | ||
| Fig. 3 (A) Fluctuation of TOF over a single 5 nm Au/TiO2 particle ([DN-BODIPY] = 2.0 μM, IUV = 0.5 W cm−2). (B) Distribution of <TOF> and its RSD over 10 different individual particles for TiO2 (black), 5 nm Au/TiO2 (blue), 8 nm Au/TiO2 (green), and 14 nm Au/TiO2 (red). | ||
Fig. 4A and 4D show the trajectories of the fluorescence intensity obtained for single TiO2 and 14 nm Au/TiO2 particles in Ar-saturated DN-BODIPY solution (2.0 μM in methanol) under 488 nm laser (I488 = 0.1 kW cm−2) and UV irradiation (IUV = 0.5 W cm−2), respectively. Because the fluorescent spots from individual bursts are spatially distributed on the surface of the particle (Fig. 4B and 4E, see the corresponding colors in panels A and D, respectively), the locations of the fluorescence bursts were determined with an accuracy of about 20–30 nm by employing a two-dimensional Gaussian distribution.13,14 Interestingly, as shown in Fig. 4C and 4F, when compared to those for a single TiO2 particle (over 5 centers), a single 14 nm Au/TiO2 particle had a limited number of reactive centers (2–3 centers) (see red circles in panel F), being equal to the number of Au nanoparticles loaded onto one TiO2 particle. A similar tendency was observed when over 10 individual particles were examined. These results suggest that the reduction of DN-BODIPY over Au/TiO2 occurs more easily on the surface of Au nanoparticles than unmodified TiO2 surfaces.
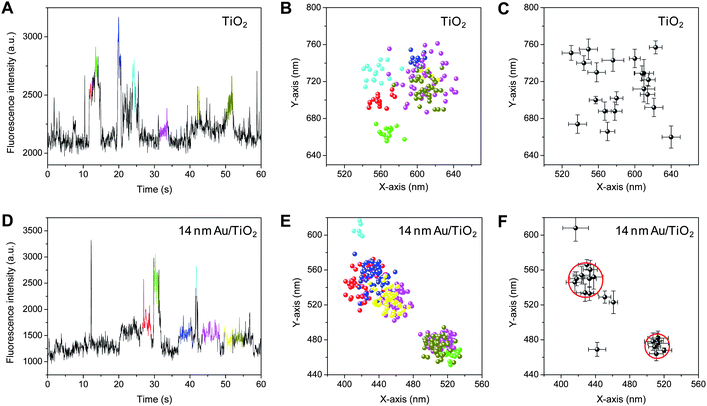 | ||
| Fig. 4 (A, D) Fluorescence intensity trajectories obtained for single TiO2 (A) and 14 nm Au/TiO2 (D) particles in Ar-saturated DN-BODIPY solution (2.0 μM, in methanol) under 488 nm laser (I488 = 0.1 kW cm−2) and UV irradiation (IUV = 0.5 W cm−2). (B, E) The locations of fluorescence bursts determined using centroid analysis of each fluorescent spot obtained for single TiO2 (B) and 14 nm Au/TiO2 (E) particles. The integration time per frame was 50 ms. See the corresponding colors in panels A and D for panels B and E, respectively. (C, F) The spatial distribution of reactive sites determined by fitting a two-dimensional Gaussian function to the fluorescence spot distributions (dots in panels C and E) collected from identical single TiO2 (C) and 14 nm Au/TiO2 (F) particles. Note that the locations of the burst with a short duration, i.e., a small number of images, were determined by centroid analysis. The red circles in panel F indicate the highly active reaction centers. The particle size of TiO2 is about 150 nm. | ||
[S]-Dependence of τoff and τon
To clarify the fundamental principles governing the photocatalytic activity of Au/TiO2, we examined the substrate concentration ([S])-dependent photocatalytic behavior at the fixed UV light intensity (IUV) of 0.5 W cm−2. In the single-particle fluorescence intensity trajectory (Fig. 5A), the actual photocatalytic events could be separated into two characteristic durations, τoff and τon, where τoff is the characteristic time prior to the formation of fluorescence products on the TiO2 or Au/TiO2 and τon is the characteristic time for which persistent emission is exhibited, which might be related to the consecutive reduction or dissociation of fluorescent products. As shown in Fig. 5B and 5C, respectively, the distributions of τon and τoff are well fitted with a single-exponential decay function (R2 > 0.97). A noticeable deviation at longer on time (e.g., τon > 4 s in Fig. 5C) would be partially due to the multiple burst events.![(A) A typical fluorescence intensity trajectory observed for a single 8 nm Au/TiO2 particle in an Ar-saturated DN-BODIPY solution (2.0 μM, in methanol) under 488 nm laser and UV light irradiation. The corresponding fluorescence intensity histogram is shown in the right panel. The green solid and dashed lines indicate the Gaussian-fitted background and the threshold level separating the on and off states, respectively, which corresponds to 3σ, where σ is the standard deviation. (B) Off and (C) on time distributions constructed from over 150 events for 20 different single 8 nm Au/TiO2 particles ([DN-BODIPY] = 2.0 μM, IUV = 0.5 W cm−2). (D, E) DN-BODIPY concentration dependence of <τoff> (D) and <τon> (E) obtained for TiO2 (black), 5 nm Au/TiO2 (blue), 8 nm Au/TiO2 (green), and 14 nm Au/TiO2 (red). The solid lines in panel D were obtained from eqn (1), and the fitting parameters are summarized in Table 1. The solid lines in panel E are guides for the eye.](/image/article/2011/SC/c0sc00648c/c0sc00648c-f5.gif) | ||
| Fig. 5 (A) A typical fluorescence intensity trajectory observed for a single 8 nm Au/TiO2 particle in an Ar-saturated DN-BODIPY solution (2.0 μM, in methanol) under 488 nm laser and UV light irradiation. The corresponding fluorescence intensity histogram is shown in the right panel. The green solid and dashed lines indicate the Gaussian-fitted background and the threshold level separating the on and off states, respectively, which corresponds to 3σ, where σ is the standard deviation. (B) Off and (C) on time distributions constructed from over 150 events for 20 different single 8 nm Au/TiO2 particles ([DN-BODIPY] = 2.0 μM, IUV = 0.5 W cm−2). (D, E) DN-BODIPY concentration dependence of <τoff> (D) and <τon> (E) obtained for TiO2 (black), 5 nm Au/TiO2 (blue), 8 nm Au/TiO2 (green), and 14 nm Au/TiO2 (red). The solid lines in panel D were obtained from eqn (1), and the fitting parameters are summarized in Table 1. The solid lines in panel E are guides for the eye. | ||
The inverse of the average values of τoff (<τoff>) and τon (<τon>) are plotted against [S] (Fig. 5D). The values of <τoff>−1 over all Au/TiO2 samples were found to be much larger than those over bare TiO2 particles under all tested substrate concentrations. This result is qualitatively consistent with the bulk measurements, in which Au/TiO2 exhibited excellent photocatalytic activity when compared to the bare TiO2 (Fig. S2†). Moreover, the <τoff>−1 values are strongly dependent on [S]. At higher concentrations, adsorption equilibrium is attained more quickly, which facilitates the reduction of DN-BODIPY to the fluorescent products, leading to enhancement of <τoff>−1. The [S]-dependence of the product formation rate can be described by the Langmuir–Hinshelwood equation:16,32
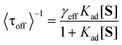 | (1) |
Interrelated IUV- and dAu-dependence of τoff and τon
The effects of IUV on τoff and τon are illustrated in Fig. 6A and 6B, respectively. Because <τoff>−1 is proportional to the total number of catalytic sites on each Au/TiO2 particle, which is related to both NAu and the surface area of one Au particle (AAu), the catalytic reactivity per Au nanoparticle per surface area (<τoff>s−1, s−1 particle−1 nm−2) can be used to evaluate the size-dependent activity of single Au/TiO2 particles: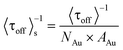 | (2) |
![UV light intensity dependence of <τoff>s−1 (A) and <τon> (B) obtained for 5 nm Au/TiO2 (blue), 8 nm Au/TiO2 (green), and 14 nm Au/TiO2 (red) ([DN-BODIPY] = 2.0 μM).](/image/article/2011/SC/c0sc00648c/c0sc00648c-f6.gif) | ||
| Fig. 6 UV light intensity dependence of <τoff>s−1 (A) and <τon> (B) obtained for 5 nm Au/TiO2 (blue), 8 nm Au/TiO2 (green), and 14 nm Au/TiO2 (red) ([DN-BODIPY] = 2.0 μM). | ||
It was found that increasing IUV leads to an increase in <τoff>s−1 owing to the accelerated formation of photogenerated electrons in TiO2 and enhanced sequential ET from TiO2 to Au (Fig. 6A). In addition, <τoff>s−1 increased as dAu decreased at a certain value of IUV. For example, <τoff>s−1 of 5 nm Au/TiO2 was 1.95 times higher than that of 14 nm Au/TiO2 at an IUV of 0.015 W cm−2. However, this difference in <τoff>s−1 between 5 nm and 14 nm Au/TiO2 particles gradually decreased to 1.03 times as the IUV increased to 0.5 W cm−2.
It is interesting to note that, except for 5 nm Au/TiO2, IUV affects the time required for HN-BODIPY to disappear from the Au/TiO2 surface (Fig. 6B). In particular, the τon value for the 14 nm Au/TiO2 sample greatly increased from 0.58 ± 0.02 s to 1.0 ± 0.1 s as IUV increased from 0.015 W cm−2 to 0.5 W cm−2. If the duration of the on time is ascribed to the consecutive reduction of fluorescent HN-BODIPY to non-fluorescent reduced species, i.e., 4-amino-3-nitrophenyl-BODIPY,21 the excitation of Au/TiO2 with weak UV light should prolong the on time of the fluorescence bursts. Because this was not the case in the present study, it is possible to exclude the contribution of the HN-BODIPY reduction to the fluorescence trajectories. Furthermore, it was noted that the burst events, especially for 14 nm Au/TiO2 at strong UV power, included single HN-BODIPY molecules and the integration of multiple product molecules (e.g., the burst at 20 s in Fig. 5A). In other words, a new HN-BODIPY molecule is generated before earlier ones dissociate away from the particle surface. Such multi-level on events directly reflect a multitude of either reduction sites that can undertake photocatalysis in parallel or adsorption sites at which products can remain on the surface before dissociation. The difference between <TOF> and <τoff>−1 is thus attributed to the multiple burst events involved in the single-molecule turnover trajectory. Because no apparent changes in the photocatalytic activity and size of the Au nanoparticles were observed during photoirradiation (Fig. S4†), the number of adsorption sites would not be affected by UV power. In contrast, the effective reduction sites, which are related to the accumulated electron concentration in Au/TiO2, may be controlled by IUV and the electron storage ability of Au particles. Larger Au nanoparticles can store more electrons (Table S1†), and the stronger excitation intensity can induce more electrons to transfer from TiO2 to Au, both of which would increase the probability of multiple parallel reduction reactions in Au nanoparticles, thereby leading to longer τon values. Accordingly, the proportion of bursts with multiple levels to the total number of detected bursts increased from 27% to 64% when IUV increased from 0.015 W cm−2 to 0.5 W cm−2 in the 14 nm Au/TiO2 system. A striking correlation (R2 = 0.98) was also found between τon and the storage ability of Au under saturated UV excitation conditions (see Section S6 for details†). In this sense, τon cannot simply be determined by the dissociation of HN-BODIPY, but is also influenced by the number of electrons accumulated in Au nanoparticles loaded on TiO2. Overall, these findings suggest that the effects of IUV and dAu on τoff/on are intricately interrelated and that the photocatalytic behavior of TiO2 loaded with larger Au nanoparticles is more dependent on IUV when compared to smaller Au nanoparticles.
Kinetic mechanism of Au charging-controlled photocatalytic reduction
Based on the above experimental results, we propose a possible mechanism for the photocatalytic reduction of DN-BODIPY over Au/TiO2, as illustrated in Fig. 7. The results confirmed that the DN-BODIPY molecules prefer to be adsorbed on the Au surface rather than the TiO2 surface (Fig. 5D and Table 1), and their reduction likely occurs on Au nanoparticles (Fig. 4). The accumulation of electrons in the Au nanoparticles increases their EF* step by step until the EF* exceeds the reduction potential of DN-BODIPY (namely, the charging process of Au),8–10 after which the electron flow from the charged Au nanoparticles to adsorbed DN-BODIPY molecules leads to the formation of HN-BODIPY concomitantly with turning on fluorescence. The reduction of single or multiple DN-BODIPY molecules adsorbed on the surface eventually depletes the accumulated electrons in the Au, which is followed by a decrease in EF* (namely, the discharging process of Au). This decrease in EF* is a preceding event before the transition to the off state.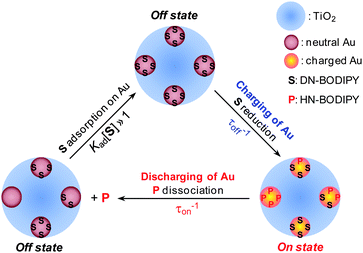 | ||
| Fig. 7 Schematic of kinetic mechanism for photocatalytic reduction over Au/TiO2. S and P are the substrate (DN-BODIPY) and fluorescent product (HN-BODIPY), respectively. The blue, brown, and orange spheres show the TiO2, and the neutral and charged Au nanoparticles, respectively. The charging process occurs during the τoff until the EF of a Au nanoparticle raises to a certain level, at which a product is generated and discharging occurs. This event marks the end of τoff and the start of τon. Discharging continues during τon to generate more product molecules until the EF drops to a certain level; the τon does not end, however, until all product molecules dissociate from the nanoparticle surface. | ||
To better understand the charging-discharging process of Au nanoparticles influencing τoff and τon, we developed a kinetic model that describes the Au charging-controlled photocatalytic reduction of DN-BODIPY over single Au/TiO2 particles under substrate-saturating conditions (i.e., Kad[S] > 1). It should be noted that once any one of Au nanoparticles are changed, photocatalytic reduction of DN-BODIPY is triggered. Consequently, ET through nanocomposite interfaces is resolved into that between single TiO2 nanoparticles and several individual Au nanoparticles. In light of the photochemical equilibrium state, the following expression for the corresponding reduction in the rate of DN-BODIPY (<τoff>s−1) under light-rich conditions was obtained:
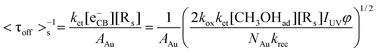 | (3) |
Eqn (3) clearly represents a strong dependence of <τoff>s−1 on AAu, NAu, [Rs], and IUV. Specifically, the charging process of Au nanoparticles should be among the most fundamental factors controlling the ET characteristics, which provides the dependence of <τoff>s−1 on IUV with a predicted power coefficient of 0.5 through [eCB−]. The linear and square root dependences of the reaction rate on light intensity in TiO2 photocatalysis have been observed by many research groups. Although the reaction rate is independent of light intensity, it is limited by mass transport at very high light intensities.36 As expected, <τoff>s−1 is dependent on IUV and the correlation coefficients of the regression straight lines indicate a satisfactory linearity between log(<τoff>s−1) and log(IUV) (R2 > 0.90) as shown in Fig. 6A. Nevertheless, the obtained slope values (n) were considerably lower than the predicted limit (n = 0.5), implying that the reduction of DN-BODIPY is controlled not only by the photons, but also by the mass transfer. Similar phenomena have been observed during the photocatalytic degradation of oxalic acid on the TiO2 layer and the photostimulated adsorption of oxygen on ZrO2.37,38 Nevertheless, the extent of IUV-dependence of <τoff>s−1 (i.e., n) differed notably among the three different sized Au/TiO2 samples, being 0.06, 0.11, and 0.24 for 5 nm, 8 nm, and 14 nm Au/TiO2, respectively (Table 1). In fact, it has reported that the rate of the photocatalyzed reaction can also be described by eqn (4), and that the power coefficient n varies with substrate concentration (c), approaching unity as c → ∞ and conversely n → 0 if c → 0.38,39
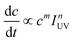 | (4) |
It has also been shown that n is related to the substrate flow rate or oxygen flux, and that enhancement of the photocatalysis rate by IUV increases as the oxygen concentration or substrate flow increases.37 According to the model of diffusion-limited kinetics developed by Roeffaers and co-workers,14 however, it is likely that there is no significant depletion of substrate from the bulk solution by catalysis under the present experimental conditions.
Similarly, <τoff>s−1 is greatly limited by the interfacial ET process between TiO2 and Au nanoparticles (i.e., ket[eCB−][Rs]) under substrate-saturating conditions; thus, it is reasonable to assume that the interdependence of <τoff>s−1 on IUV and dAu primarily arises from the different electron storage ability of Au nanoparticles as shown in eqn (5):
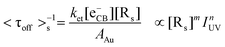 | (5) |
As dAu increases from 5.5 to 14.1 nm, AAu increases from 95.0 to 624.3 nm2, and the electron storage ability of Au nanoparticles (q), which increases in proportion to AAu,40 is enlarged significantly from 154 to 1014 electrons particle−1 (Section S6†). That is, [Rs] increased from 154 to 1014 sites per Au nanoparticle. Since the number of surface trapping sites for electrons on 5 nm Au is relatively limited, additional photon flux could not efficiently increase the rate of ET from TiO2 CB to Au nanoparticles; accordingly, n approaches zero (0.06). In contrast, 14 nm Au nanoparticles provide many more surface reactive sites, which results in their photocatalytic activity being more sensitive to IUV (n = 0.24). This interpretation is further supported by the positive correlation between n and q (R2 = 0.98, Fig. S6†).
Efficiency of photocatalytic reduction on Au/TiO2
In terms of multiple molecules involving-on events, τon reflects the concentration of electrons temporarily accumulated in Au nanoparticles, and thus should be combined with τoff to assess the photocatalytic ability of Au/TiO2. Here, we define the <τon>/<τoff> (being equal to <τon>s/<τoff>s) obtained under light-controlled conditions (i.e., Kad[S] > 1) as the single-particle activity factor to rationally evaluate the inherent photocatalytic activity for the reduction over individual Au/TiO2 particles. Since <τoff>−1 represents the formation rate of HN-BODIPY and <τon> is proportional to the number of simultaneously generated HN-BODIPY molecules, the values of <τon>/<τoff> can reflect the rough amount of produced HN-BODIPY per unit of time.Fig. 8 illustrates the relationships between dAu, IUV, and <τon>/<τoff>. The most interesting finding is the opposite dependence of the photocatalytic activity on dAu that was observed at different IUV, which makes the reaction mechanism more complicated. Although a similar contradiction was observed in previous studies,9,10 their proposed mechanisms in terms of the quantum size and solvent polarity effects on EF* cannot fully explain our results. We believe that our findings originate from the size-controlled charging process of the Au nanoparticles leading to the interdependence of τoff/on on IUV and dAu. Because smaller Au nanoparticles require substantially fewer electrons to raise their EF, the charging process can be accomplished rapidly, even upon weak UV excitation, while the charging of larger Au nanoparticles would be greatly limited because of the relatively insufficient photogenerated electrons in TiO2. Such superiority causes Au/TiO2 particles with smaller dAu values to show better photocatalytic activity at low IUV. Simultaneously, the limited surface trapping sites for electrons on smaller Au particles results in the photocatalytic behavior of 5 nm Au/TiO2 being less dependent on IUV than Au/TiO2 with larger dAu values, as discussed above. However, intense UV excitation can produce excess electrons in TiO2 to efficiently induce Au charging; hence, it greatly reduces the difference in τoff for Au/TiO2 with various dAu (Fig. 6A). Once the reaction attains the new equilibrium EF* state, larger Au nanoparticles would shuttle more electrons to reduce multiple DN-BODIPY molecules at one time, leading to a greatly prolonged τon (Fig. 6B). Therefore, the rapid charging process accompanied with an extended discharging time will cause enhanced photocatalytic activity for Au/TiO2 with increasing dAu at high IUV.
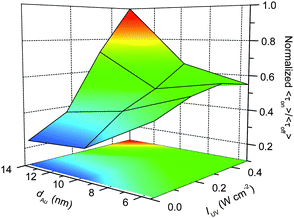 | ||
| Fig. 8 d Au and IUV dependence on <τon>/<τoff> values obtained for 5, 8, and 14 nm Au/TiO2. | ||
Conclusions
In conclusion, we investigated the photocatalytic redox reaction of the dinitrobenzene moiety over single TiO2 or Au/TiO2 particles using single-molecule fluorescence imaging techniques with a redox-responsive fluorescent probe, DN-BODIPY. Examination of the location of fluorescent spots emitted from the products using super-resolution fluorescence microscopy with single molecule sensitivity allowed us to map the distribution of the reactive centers on the surface of the particles. All of the tested Au/TiO2 samples showed much higher photocatalytic reduction ability than TiO2, because the loaded Au nanoparticles greatly enhance the charge separation within the nanocomposites. Statistical analysis of the single-molecule TOFs revealed that the Au/TiO2 particles exhibit smaller activity fluctuation when compared to bare TiO2. The dependence of fluorescence on and off times (τon and τoff) on the substrate concentration, UV light intensity, and size of Au nanoparticles was also examined and evaluated in terms of the Au charging-controlled reaction mechanism developed in this study. Our single-molecule, single-particle approach provides further insight into the mechanism by which (photo)catalytic reactions at a heterogeneous interface occur, as well as the transport characteristics of electrons in composite systems consisting of semiconductors, metals, and organic compounds.Experimental section
Synthesis of DN-BODIPY
8-(3,4-Dinitrophenyl)-1,3,5,7-tetramethyl-4,4-difluoro-4-bora-3a,4a-diaza-s-indacene (DN-BODIPY) was synthesized as previously reported.21Preparation and characterization of Au/TiO2
A series of Au/TiO2 particles (5, 8, and 14 nm Au/TiO2) were prepared by the deposition-precipitation method under various temperatures and times using HAuCl4 and TiO2 (Ishihara Sangyo, A-100, 100–200 nm particles) as the raw materials.41 Detailed descriptions of the morphology (TEM images), crystal structure (powder XRD pattern), and optical properties (steady-state UV-visible diffuse reflectance spectra) of the synthesized Au/TiO2 particles are given in the Supporting Information (Fig. S1†).Ensemble-averaged spectral measurements
Steady-state UV–visible absorption and diffuse reflectance spectra were measured using UV–visible–NIR spectrophotometers (Shimadzu UV-3100 and Jasco V-570, respectively). Steady-state fluorescence spectra were measured using a Hitachi 850 fluorescence spectrophotometer.Single-molecule fluorescence measurements by TIRFM
The experimental setup was based on an Olympus IX71 inverted fluorescence microscope. The details of the experimental setup are described in the Supporting Information†. To obtain isolated TiO2 or Au/TiO2 particles, well-dispersed methanol suspensions containing small amounts of TiO2 or Au/TiO2 powders were spin-coated onto clean cover glasses. The surface density of the particles on the cover glasses was very low, with only one particle being present in a 12.5 μm × 12.5 μm area. The position of the TiO2 or Au/TiO2 particles immobilized on the cover glass was determined from the transmission image obtained by illuminating the samples using a halogen lamp (Olympus, U-LH100L-3). A circular-polarized light emitted from a CW Ar ion laser (Melles Griot, IMA101010BOS; 488 nm, 0.1 kW cm−2 at the glass surface) was reflected toward a second dichroic mirror (Olympus, DM505) using a first dichroic mirror (Olympus, RDM450), which reflected wavelengths longer than 450 nm and was transparent to wavelengths shorter than 450 nm. The laser light passing through an objective lens (Olympus, UPLSAPO 100XO; 1.40 NA, 100×) after reflection by a second dichroic mirror was completely reflected at the cover glass-methanol interface. This resulted in generation of an evanescent field, which made it possible to detect a single fluorescence dye molecule. For excitation of the TiO2 or Au/TiO2 particles, the 365 nm light emitted by a LED (OPTO-LINE, MS-LED-365) and passing through an ND filter was passed through the objective. The light intensity through the objective, immersion oil, and cover glass was measured using a power meter (Ophir, Nova II) equipped with a PD300-UV head. The fluorescence emission from the fluorescent products generated over a single TiO2 or Au/TiO2 particle on the cover glass was collected using the same objective, after which it was magnified by a 1.6× built-in magnification changer, passed through a bandpass filter (Semrock, FF01-531/40-25) to remove the undesired scattered light, and then imaged using an electron-multiplying charge-coupled device (EM-CCD) camera (Roper Scientific, Cascade II:512). The images were recorded at a frame rate of 20 frames s−1 and processed using ImageJ (http://rsb.info.nih.gov/ij/) or OriginPro 8.1 (OriginLab). All experimental data were obtained at room temperature. To determine the locations of the reactive site distributed on the surface, the precise positions of the fluorescent spots were analyzed for each image using the ImageJ software with a SpotTracker plugin.42Single-molecule fluorescence measurements by confocal microscopy
Confocal fluorescence images were taken using an objective-scanning confocal microscope system (PicoQuant, MicroTime 200) coupled with an Olympus IX71 inverted fluorescence microscope. The samples were excited through an oil-immersion objective lens (Olympus, UAPON 150XOTIRF; 1.45 NA, 150×) using a 485 nm pulsed laser (PicoQuant, LDH-D-C-485) controlled by a PDL-800B driver (PicoQuant). The emission from the sample was collected using the same objective and detected by a single photon avalanche photodiode (Micro Photon Devices, PDM 50CT) through a dichroic beam splitter and bandpass filter (Semrock, FF01-531/40-25). All experimental data were obtained at room temperature. The details of the experimental setup and procedures are described in the Supporting Information†.Acknowledgements
N. W. gives thanks to Prof. Lihua Zhu at Huazhong University of Science and Technology and the CSC (China Scholarship Council) program for their support. T. M. gives thanks to the WCU (World Class University) program through the National Research Foundation of Korea funded by the Ministry of Education, Science and Technology (R31-10035) for their support. This work has been partly supported by a Grant-in-Aid for Scientific Research (Projects 22245022, 21750145, and others) from the Ministry of Education, Culture, Sports, Science and Technology (MEXT) of the Japanese Government.References
- M. Grätzel and J. E. Moser, Electron Transfer in Chemistry, Vol. 5, Wiley-VCH, Weinheim, 2001 Search PubMed.
- P. V. Kamat, J. Phys. Chem. C, 2007, 111, 2834–2860 CrossRef CAS.
- P. V. Kamat, J. Phys. Chem. C, 2008, 112, 18737–18753 CAS.
- A. Corma and H. Garcia, Chem. Soc. Rev., 2008, 37, 2096–2126 RSC.
- H. Tada, T. Kiyonaga and S. Naya, Chem. Soc. Rev., 2009, 38, 1849–1858 RSC.
- D. V. Talapin, J.-S. Lee, M. V. Kovalenko and E. V. Shevchenko, Chem. Rev., 2010, 110, 389–458 CrossRef CAS.
- Y. Nakato, M. Shioji and H. Tsubomura, Chem. Phys. Lett., 1982, 90, 453–456 CrossRef CAS.
- V. Subramanian, E. Wolf and P. V. Kamat, J. Phys. Chem. B, 2001, 105, 11439–11446 CrossRef CAS.
- V. Subramanian, E. E. Wolf and P. V. Kamat, J. Am. Chem. Soc., 2004, 126, 4943–4950 CrossRef CAS.
- T. Kiyonaga, M. Fujii, T. Akita, H. Kobayashi and H. Tada, Phys. Chem. Chem. Phys., 2008, 10, 6553–6561 RSC.
- B. M. Weckhuysen, Angew. Chem., Int. Ed., 2009, 48, 4910–4943 CrossRef CAS.
- M. B. J. Roeffaers, B. F. Sels, H. Uji-i, F. C. De Schryver, P. A. Jacobs, D. E. De Vos and J. Hofkens, Nature, 2006, 439, 572–575 CrossRef CAS.
- M. B. J. Roeffaers, G. De Cremer, J. Libeert, R. Ameloot, P. Dedecker, A.-J. Bons, M. Bueckins, J. A. Martens, B. F. Sels, D. E. De Vos and J. Hofkens, Angew. Chem., Int. Ed., 2009, 48, 9285–9289 CrossRef CAS.
- G. De Cremer, M. B. J. Roeffaers, E. Bartholomeeusen, K. Lin, P. Dedecker, P. P. Pescarmona, P. A. Jacobs, D. E. De Vos, J. Hofkens and B. F. Sels, Angew. Chem. Int. Ed., 2010, 49, 908–911 CAS.
- G. De Cremer, B. F. Sels, D. E. De Vos, J. Hofkens and M. B. J. Roeffaers, Chem. Soc. Rev., 2010, 39, 4703–4717 RSC.
- W. Xu, J. S. Kong, Y.-T. E. Yeh and P. Chen, Nat. Mater., 2008, 7, 992–996 CrossRef CAS.
- W. Xu, H. Shen, Y. J. Kim, X. Zhou, G. Liu, J. Park and P. Chen, Nano Lett., 2009, 9, 3968–3973 CrossRef CAS.
- X. Zhou, W. Xu, G. Liu, D. Panda and P. Chen, J. Am. Chem. Soc., 2010, 132, 138–146 CrossRef CAS.
- K. Naito, T. Tachikawa, M. Fujitsuka and T. Majima, J. Am. Chem. Soc., 2009, 131, 934–936 CrossRef CAS.
- T. Tachikawa and T. Majima, J. Am. Chem. Soc., 2009, 131, 8485–8495 CrossRef CAS.
- T. Tachikawa, N. Wang, S. Yamashita, S.-C. Cui and T. Majima, Angew. Chem., Int. Ed., 2010, 49, 8593–8597 CrossRef CAS.
- T. Tachikawa and T. Majima, Chem. Soc. Rev., 2010, 39, 4802–4819 RSC.
- P. Gao, D. Gosztola and M. J. Weaver, J. Phys. Chem., 1988, 92, 7122–7130 CrossRef CAS.
- A. Corma, P. Concepcion and P. Serna, Angew. Chem., Int. Ed., 2007, 46, 7266–7269 CrossRef CAS.
- A. Loudet and K. Burgess, Chem. Rev., 2007, 107, 4891–4932 CrossRef CAS.
- T. Ueno, Y. Urano, H. Kojima and T. Nagano, J. Am. Chem. Soc., 2006, 128, 10640–10641 CrossRef CAS.
- C.-Y. Wang, J. Rabani, D. W. Bahnemann and J. K. Dohrmann, J. Photochem. Photobiol., A, 2002, 148, 169–176 CrossRef CAS.
- A. A. Ismail, D. W. Bahnemann, I. Bannat and M. Wark, J. Phys. Chem. C, 2009, 113, 7429–7435 CrossRef CAS.
- S. A. Haque, Y. Tachibana, R. L. Willis, J. E. Moser, M. Grätzel, D. R. Klug and J. R. Durrant, J. Phys. Chem. B, 2000, 104, 538–547 CrossRef CAS.
- J. Nelson and R. E. Chandler, Coord. Chem. Rev., 2004, 248, 1181–1194 CrossRef CAS.
- W. R. Duncan and O. V. Prezhdo, Annu. Rev. Phys. Chem., 2007, 58, 143–184 CrossRef CAS.
- W. Xu, J. S. Kong and P. Chen, J. Phys. Chem. C, 2009, 113, 2393–2404 CrossRef CAS.
- J. L. Ferry and W. H. Glaze, J. Phys. Chem. B, 1998, 102, 2239–2244 CrossRef CAS.
- R. Van Hardeveld and F. Hartog, Surf. Sci., 1969, 15, 189–230 CrossRef CAS.
- H. Tada, T. Ishida, A. Takao, S. Ito, S. Mukhopadhyay, T. Akita, K. Tanaka and H. Kobayashi, ChemPhysChem, 2005, 6, 1537–1543 CrossRef CAS.
- D. F. Ollis, E. Pelizzetti and N. Serpone, Environ. Sci. Technol., 1991, 25, 1522–1529.
- J. Krysa, L. Vodehnal and J. Jirkovsky, J. Appl. Electrochem., 1999, 29, 429–435 CrossRef CAS.
- A. V. Emeline, A. V. Rudakova, V. K. Ryabchuk and N. Serpone, J. Phys. Chem. B, 1998, 102, 10906–10916 CrossRef CAS.
- A. V. Emeline, V. Ryabchuk and N. Serpone, J. Photochem. Photobiol., A, 2000, 133, 89–97 CrossRef CAS.
- A. Toyota and T. Sagara, Electrochim. Acta, 2008, 53, 2553–2559 CrossRef CAS.
- T. Kiyonaga, T. Mitsui, M. Torikoshi, M. Takekawa, T. Soejima and H. Tada, J. Phys. Chem. B, 2006, 110, 10771–10778 CrossRef CAS.
- D. Sage, R. Neumann Franck, F. Hediger, M. Gasser Susan and M. Unser, IEEE Trans Image Process, 2005, 14, 1372–1383 CrossRef.
Footnote |
| † Electronic Supplementary Information (ESI) available: Movie showing the single-molecule, single-particle fluorescence imaging of the reduction of DN-BODIPY on 8 nm Au/TiO2 (Movie S1). Experimental details including the descriptions of the movie (S0), the experimental methods (S1), the preparation and characterization of Au/TiO2 (S2), the ensemble-averaged photocatalytic activity (S3), the turnover rate of off-on cycle (S4), the influence of photoirradiation on the Au particle size (S5), the size dependent of electron storage ability of Au nanoparticle (S6), and the kinetic analysis of Au/TiO2 photocatalytic reactions (S7). See DOI: 10.1039/c0sc00648c/ |
| This journal is © The Royal Society of Chemistry 2011 |