Active and reusable Pd(II) organometallic catalyst covalently bonded to mesoporous silica nanospheres for water-medium organic reactions†
Wenhan
He
,
Fang
Zhang
and
Hexing
Li
*
The Education Ministry Key Lab. of Res. Chem., Shanghai Normal University, Shanghai, 200234, P.R. China. E-mail: Hexing-Li@shnu.edu.cn; Fax: +021-64322272; Tel: +021-64322272
First published on 15th February 2011
Abstract
A novel heterogenized Pd(II) organometallic catalyst was synthesized by coordinating Pd(II) ions with PPh2-functionalized silica nanospheres containing an ordered mesoporous structure. This catalyst exhibited higher activity and selectivity in a water-medium for the Barbier, Sonogashira and Ullmann reactions than the corresponding catalyst bonded to MCM-41. This was found to be due to high surface area, ordered mesopores and short pore channels, which facilitated the diffusion and adsorption of reactant molecules. This catalyst displayed comparable efficiencies with the homogeneous catalyst, Pd(PPh2)3Cl2 and could be easily recycled and used repetitively. It showed good potential in industrial applications owing to the reduced cost and the diminished environmental pollution from either the organic solvents as reaction media or the heavy metallic ions.
Introduction
Water-medium organic reactions have received increasing interest since water is the safest and cheapest solvent in the world. Most studies are focused on homogeneous catalysts.1,2 Despite high activity and selectivity, their industrial applications are still limited by difficult separation and reuse, which eventually adds cost and even causes environmental pollution from heavy metallic ions.3 Heterogeneous catalysts can be easily recycled, but they usually exhibit poor activity due to the low dispersion degree of active sites and an enhanced diffusion limit.4–6 Previously, we reported a series of organometallic catalysts bonded to silica supports with ordered mesoporous channels, such as MCM-41 and SBA-15 etc., which showed significant improvement on the adsorption and diffusion of reactant molecules, leading to the enhanced activity.7,8 However, their irregular shape and long pore channels are harmful to their catalytic performances due to an enhanced diffusion limit. Recently, mesoporous silica nanospheres (MSN) with an average diameter around 100 nm have been reported.9 Obviously, such silica supports should be more favorable for the diffusion and adsorption of reactant molecules owing to their short pore channels and their regular nanospheres. In this paper, we report for the first time the synthesis of a novel Pd(II) organometallic catalyst covalently bonded to PPh2-functionalized silica nanospheres with ordered mesopores. This catalyst exhibited higher activity and selectivity in water-medium C–C coupling reactions than the same catalyst bonded to MCM-41. The catalyst displayed comparable efficiencies with the traditional homogeneous catalyst Pd(PPh2)3Cl2 and could be used repeatedly.Experimental
Catalyst preparation
Firstly, the PPh2-functionalized silica nanospheres with ordered mesoporous channels, denoted as PPh2-MSN, were synthesized by surfactant (CTAB)-directed co-condensation between tetraethyl silicate (TEOS) and PPh2CH2CH2Si(OEt)3 under basic condition. Briefly, 0.20 g CTAB was dissolved in 96 ml H2O containing 1.4 mmol NaOH at 80 °C. After being stirred for 1.5 h, the total Si amount from both TEOS and PPh2CH2CH2Si(OEt)3 was fixed at 1.0 mmol. After being stirred for 2.0 h, the mixture was kept static for another 20 h. The white precipitate was filtered and washed thoroughly with water, followed by vacuum drying at 80 °C overnight. The surfactant and other organic residues were then removed by refluxing in 500 ml ethanol solution containing 0.50 M HCl at 80 °C for 24 h, leading to pure PPh2-MSN. By adjusting the PPh2CH2CH2Si(OEt)3/TEOS molar ratio to 5.0%, 8.0% and 11%, three PPh2-MSN samples (PPh2-MSN-1, PPh2-MSN-2, and PPh2-MSN-3) were obtained, corresponding to the theoretical PPh2/Si molar ratios of 4.7%, 7.4% and 9.9%, respectively. Then, the as-received PPh2-MSN sample was dispersed in 5.0 ml anhydrous toluene containing Pd(PPh3)2Cl2 and was kept stirring at 25 °C for 24 h. The yellow solid was filtrated and washed with toluene and CH2Cl2, followed by vacuum drying at 80 °C overnight, leading to the Pd-PPh2-MSN catalyst. The Pd-loading could be adjusted by changing the Pd(PPh3)2Cl2 content in the mother mixture.For comparison, the catalyst Pd-PPh2-MCM-41 was also synthesized according to the following procedures. Firstly, 0.18 g CTAB was dissolved in 12 ml H2O containing 7.5 g NH3·H2O (28 wt%) at 40 °C, followed by simultaneously adding 1.6 ml TEOS and 0.21 ml PPh2CH2CH2Si(OEt)3. After being stirred for 1.5 h, the mixture was transferred into an autoclave and kept at 100 °C for 15 h. The white precipitate was filtered and washed thoroughly with water, followed by vacuum drying at 80 °C overnight. The surfactant and other organic residues were then removed by refluxing 500 ml ethanol solution containing 0.50 M HCl at 80 °C for 24 h. The as-prepared PPh2-functionalized silica, with a mesoporous structure similar to the MCM-41, was denoted as PPh2-MCM-41. Then, the Pd-PPh2-MCM-41 was prepared by reacting PPh2-MCM-41 with Pd(PPh3)2Cl2 according to the method described above.
Characterization
The metal loadings were determined by an inductively coupled plasma optical emission spectrometer (ICP, Varian VISTA-MPX). Solid state nuclear magnetic resonance (NMR) spectra were recorded on a Bruker AV-400 spectrometer. Fourier transform infrared (FTIR) spectra were collected on a Nicolet Magna 550 spectrometer by using KBr method. Thermogravimetric analysis and differential thermal analysis (TG/DTA) were conducted on a DT-60. X-ray powder diffraction (XRD) patterns were obtained on a Rigaku D/Max-RB diffractometer with Cu-Kα radiation. Transmission electron microscopy (TEM) images were obtained on a JEOL JEM2010. Field emission scanning electron microscopy (FESEM) pictures were observed on a Hitachi S-4800. N2 adsorption-desorption isotherms were measured at −196 °C on a Quantachrome NOVA 4000e analyzer after being degassed at 100 °C overnight. The specific surface area (SBET), pore volume (VP) and average pore diameter (DP) were calculated by applying the multiple-point Brunauer-Emmett-Teller (BET) and the Barrett-Joyner-Halenda (BJH) models based on the adsorption branches. The X-ray photoelectron spectra (XPS) were analyzed on the PHI 5000 VersaProbe. All the binding energy values were calibrated by using C1S = 284.8 eV as a reference.Adsorption test
The adsorption for RhB in aqueous solution was used as a probe to evaluate the adsorption performance of different catalysts. In a typical run of tests, 0.050 g of catalyst was added into 250 ml aqueous solution containing 20 mg L−1RhB. The solution was stirred around 200 r min−1 at 25 °C and sampled at given intervals. The RhB left in the solution was determined by UV-visiblespectrophotometry from which the adsorption capacity at different time could be calculated.Activity test
Two kinds of C–C coupling reactions (Scheme S1–2, ESI†) were employed to evaluate the catalyst performances. Water-medium Barbier reactions (S1, ESI†) were carried out at 50 °C in a 10 ml flask containing 50 μl benzaldehyde, 0.15 ml allyl bromide, 0.45 g SnCl2, 5.0 ml H2O, and a catalyst with 0.065 mmol Pd. After being refluxed for 6.0 h, the reaction mixture was extracted by toluene and dried by anhydrous MgSO4. The liquid products were quantitatively analyzed on a gas chromatograph (GC, Agilent 1790) equipped with an FID and a JWDB-5 95% dimethyl 1-(5%)-diphenylpolysiloxane column at 100 °C in N2 flow, from which both the conversion and selectivity were determined by using 2-phenylethyl as an internal standard. The conversion was calculated based on benzaldehyde since allyl bromide was excess.Water-medium Sonogashira reactions (Scheme S2, ESI†) were carried out at 80 °C in a 10 ml flask containing 0.14 ml phenyl acetylene, 0.14 ml iodobenzene, 0.21 ml DBU, 0.10 ml decane, 5.0 mg CuI, 4.0 ml H2O, and a catalyst with 0.0075 mmol Pd. After being refluxed for 2.0 h, the reaction mixture was extracted by acetic ether and dried by anhydrous MgSO4, followed by product analysis on a gas chromatograph (GC, GC-17A SHIMADZU) equipped with an FID and a JWDB-5 95% dimethyl 1-(5%)-diphenylpolysiloxane column at 80 °C in N2 flow using decane as an internal standard. The conversion was calculated based on phenyl acetylene since iodobenzene was excess. In all these experiments, the reproducibility was checked by repeating each result at least three times and was found to be within acceptable limits (±5%).
In order to determine catalyst durability, the catalyst was allowed to settle down after each run of reactions and the clear supernatant liquid was decanted. After being washed with water, acetone and ethyl ether for three times, followed by vacuum drying at 80 °C overnight, the catalyst was re-used with a fresh charge of solvent and reactant for subsequent reactions under the same conditions. After each run of reactions, the content of Pd leached off during reactions was determined by ICP analysis.
Results and discussion
The Pd loadings in different catalysts were determined by ICP analysis and are listed in the Table 1. The XPS spectra in Fig. 1 revealed that all the Pd species in the Pd-PPh2-MSN-2 were present in +2 oxidation state rather than in metallic state, corresponding to the binding energy (BE) of 343.0 and 337.8 eV in the Pd 3d5/2 and 3d3/2 levels,10 respectively. In comparison with the BE of the Pd in Pd(PPh3)2Cl2, the BE of the Pd in the Pd-PPh2-MSN-2 shifted negatively by 0.2∼0.3 eV, indicating that the PPh2-CH2-CH2- ligand exhibited stronger electron-donation than the PPh3-ligand, making the Pd atom in the Pd-PPh2-MSN more electron-enriched. This can be easily understood by considering the conjugated π–π system between P and Ph. The PPh3-ligand displayed a conjugated π–π system comprised of one P atom and three Ph groups while the PPh2-CH2-CH2-ligand exhibits a conjugated π–π system comprised of one P atom and two Ph groups. The bigger conjugated π–π system might dilute electron density on the P atom, which weakens the electron-donation ability of the P atom to the Pd atom in the coordination bonds.10| Catalyst | Pd (wt%) | S BET (m2 g−1) | D p (nm) | V p (cm3 g−1) | R | Conv. (%) | Select. (%) |
|---|---|---|---|---|---|---|---|
| a Reaction conditions: a catalyst containing 0.065 mmol Pd, 50 μl benzaldehyde, 0.15 ml allyl bromide, 0.45 g SnCl2, 5.0 ml H2O, reaction T = 50 °C, reaction time = 6.0 h. b The Pd-PPh2-MSN-2 catalyst used repeatedly 6 times. | |||||||
| Pd(PPh2)3Cl2 | — | — | — | — | H | 98 | 94 |
| Pd-PPh2-MCM-41 | 7.4 | 427 | 1.9 | 0.10 | H | 90 | 90 |
| Pd-PPh2-MSN-1 | 5.5 | 806 | 2.2 | 0.26 | H | 82 | 91 |
| Pd-PPh2-MSN-2 | 8.4 | 507 | 2.0 | 0.16 | H | 95 | 93 |
| Pd-PPh2-MSN-2 | 8.4 | 507 | 2.0 | 0.16 | Cl | 88 | 92 |
| Pd-PPh2-MSN-2 | 8.4 | 507 | 2.0 | 0.16 | CH3 | 84 | 92 |
| Pd-PPh2-MSN-3 | 12.7 | 345 | 1.8 | 0.097 | H | 77 | 92 |
| Used Pd-PPh2-MSN-2b | 8.1 | 182 | 1.6 | 0.10 | H | 80 | 90 |
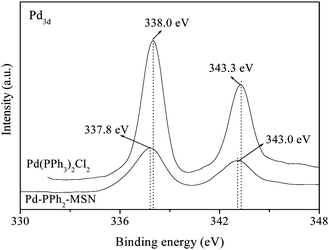 | ||
| Fig. 1 XPS spectra of Pd-PPh2-MSN-2 and Pd(PPh3)2Cl2. | ||
As shown in Fig. 2, the FTIR spectra revealed that, besides those observed in the pure SiO2, both Pd-PPh2-MSN-2 and Pd-PPh2-MCM-41 displayed new absorption peaks around 2900, 693, 3063 and 1435 cm−1, corresponding to the stretching mode of C–H bonds, the C–H out-of-plane deformation of the monosubstituted benzene ring, the C–H stretching mode of the benzene, and the vibrations of P-CH2 bond.11 These results demonstrated the successful incorporation of the PPh2 groups into the silica network.
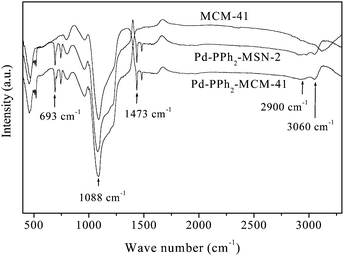 | ||
| Fig. 2 FTIR spectra of MCM-41, Pd-PPh2-MSN-2 and Pd-PPh2-MCM-41. | ||
Fig. 3 shows the solid state NMR spectra of the Pd-PPh2-MSN-2 catalyst. The 29Si CP-MAS NMR spectrum of the Pd-PPh2-MSN-2 displayed Q4 (δ = −110 ppm), Q3 (δ = −101 ppm), T3 (δ = −66.0 ppm), and T2 (δ = −58.0 ppm) signals, where Qn = Si(OSi)n(OH)4−n, n = 2–4, Tm = RSi(OSi)m(OH)3−m, m = 1–3, suggesting the presence of R–Si bonds. The 13C CP-MAS NMR spectrum revealed that Pd-PPh2-MSN-2 exhibits two peaks around 16 and 29 ppm, corresponding to the two C atoms in the –CH2–CH2– group bound with the PPh2 group. An additional peak was observed around 129 ppm due to the C atoms in the benzene ring (Ph).11 A weak broad peak around 59 ppm was assigned to the C atoms in the C2H5O- group connecting with Si due to the incomplete hydrolysis of TEOS.12 Other peaks marked with asterisks were attributed to rotational sidebands and were confirmed by changing the rotation speed.12 Besides rotational sidebands marked with asterisks, only one intense peak was observed around 20 ppm in the 31P CPMAS NMR spectrum of Pd-PPh2-MSN-2, indicating a unique coordination model between P and Pd(II).12 The 31P MAS NMR spectrum observed in the Pd-PPh2-MSN-2 catalyst was almost the same as that observed in either the PdCl2(PPh3)2 or the PdCl2[PPh2CH2Si(OCH2CH3)3]2 (Fig. S1, ESI†). The peak around 20 ppm could be assigned to the phosphor ligands in the four-coordinated Pd(II) complex, which confirmed that the Pd(II) active sites were present in monomer complexes and each Pd(II) ion coordinated with two P atoms.13 Meanwhile, the TG/DTA curves (Fig. S2, ESI†) revealed that, besides the weight loss before 200 °C due to the desorption of physically adsorbed solvents, Pd-PPh2-MSN-2 displayed a weight loss of about 30% around 378 °C, corresponding to the oxidation removal of the PPh2-CH2CH2- groups.14 There should be two kinds of Pd(II) organometals bonded to the PPh2-MSN support depending on the replacement of one PPh3 or two PPh3 in Pd(PPh3)2Cl2 by the PPh2-CH2CH2- ligands, corresponding to the theoretical weight losses around 31% and 19%, respectively. According to the experimental weight loss of 30% from TG/DTA analysis, it was reasonable to conclude that each Pd(II) coordinated with one PPh3- and one PPh2-CH2CH2- ligand in Pd-PPh2-MSN-2, rather than coordinating with two PPh2-CH2CH2- ligands.
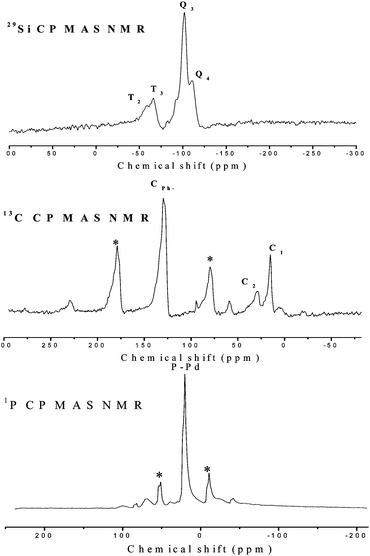 | ||
| Fig. 3 Solid NMR spectra of the Pd-PPh2-MSN-2 catalyst. | ||
The FESEM images (Fig. S3, ESI†) demonstrated that Pd-PPh2-MSN-2 was present in uniform nanospheres with an average diameter around 100 nm while Pd-PPh2-MCM-41 displayed very irregular morphologies with the average diameter bigger than 1.5 μm. The TEM images (Fig. 4) further confirmed that the Pd-PPh2-MSN was comprised of uniform nanospheres. These nanospheres contained ordered mesoporous channels similar to Pd-PPh2-MCM-41, which could also be confirmed by low-angle XRD patterns. As shown in Fig. 5, all the Pd-PPh2-MSN and Pd-PPh2-MCM-41 samples displayed a well resolved peak around 2θ of 2.1°–2.2° indicative of (100) diffraction belonging to the ordered mesoporous structure.14 Furthermore, all the Pd-PPh2-MSN samples and Pd-PPh2-MCM-41 displayed type IV N2 adsorption-desorption isotherms characteristic of mesoporous structure14 with a narrow pore size distribution around 2.0 nm (Fig. S4 and S5, ESI†).
 | ||
| Fig. 4 TEM images of Pd-PPh2-MSN-2 and Pd-PPh2-MCM-41. | ||
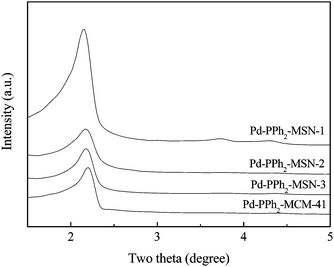 | ||
| Fig. 5 Low-angle XRD patterns of different catalysts. | ||
Other structural parameters including specific surface area (SBET), pore volume (VP) and average pore diameter (DP) are summarized in Table 1. The SBET, Dp and Vp of the Pd-PPh2-MSN samples decreased with the increase of the Pd loading due to the occupation of the Pd(II) complexes inside the pore channels. Although Pd-PPh2-MCM-41 contained lower Pd content than Pd-PPh2-MSN-2, it still exhibited lower SBET, Dp and Vp, possibly due to the poor distribution of Pd(II) complexes at the pore entrance and inside the pore channels.
Table 1 and 2 summarized the catalytic parameters of different catalysts in the water-medium Barbier reactions (Scheme 1, ESI†) and Sonogashira reactions (Scheme 2, ESI†). By adjusting the amount of catalyst added, the Pd content in the Barbier and Sonogashira reactions was fixed at 0.065 and 0.075 mmol, respectively. Pd-PPh2-MSN-2 exhibited higher activity than either Pd-PPh2-MSN-1 or Pd-PPh2-MSN-3, while the selectivity remained almost unchanged for different Pd-PPh2-MSN catalysts. At very low Pd loadings, the Pd active sites were located separately, leading to lower activity since the Barbier reaction needs two neighbouring active sites for adsorbing both the benzaldehyde and allyl bromide molecules. However, very high Pd loadings were also harmful to activity due to the damage of the mesoporous structure, leading to the lower SBET, VP and DP. For the Barbier reactions, the presence of substituent group (R) on the benzaldehyde had no significant influence on the selectivity but caused a considerable decrease in the activity regardless of the electron-withdrawing or the electron-donating property, possibly due to the enhanced steric hindrance. For the Sonogashira reactions, the presence of substitute (R) on the iodobenzene also disfavored activity. However, it was clearly found that electron-donating substituents were more harmful than electron withdrawing substituents, because electron withdrawing substituents make the carbon of the C–I bond more electropositive which favors the Sonogashira reaction.15Pd-PPh2-MSN-2 displayed almost the same activity as the traditional Pd(PPh3)2Cl2 homogeneous catalyst in both the Barbier or Sonogashira reactions. To establish whether the heterogeneous or the dissolved homogeneous Pd(II) species was the real catalyst responsible for the Barbier reaction, the following procedure was carried out according the procedure proposed by Sheldon et al.16 After reacting for 6 h with the conversion exceeding 30%, the solid catalyst was filtered and the mother liquor was allowed to react for another 12 h under the same conditions. No significant change in either the conversion or the product yield was detected, indicating that the reaction completely stopped from which one could conclude that the active phase was not dissolved Pd(II) species leached from the supports. Therefore, the present catalysis was really heterogeneous in nature.
| Catalyst | Additive | R | Conversion (%) | Selectivity (%) | Yield (%) |
|---|---|---|---|---|---|
| a Reaction conditions: catalyst containing 0.0075 mmol Pd, 0.14 ml phenyl acetylene, 0.14 ml iodobenzene, 0.12 ml DBU, 0.10 ml heptane, 5.0 mg CuI or 3.7 mg CuBr, 4.0 ml H2O, reaction T = 80 °C, reaction time = 2.0 h. | |||||
| Pd-PPh2-MSN-2 | CuI | H | 98 | 98 | 97 |
| Pd-PPh2-MSN-2 | CuI | NO2 | 96 | 98 | 95 |
| Pd-PPh2-MSN-2 | CuI | CH3 | 94 | 96 | 90 |
| Pd-PPh2-MSN-2 | CuI | OCH3 | 90 | 98 | 88 |
| Pd-PPh2-MCM-41 | CuI | H | 94 | 90 | 85 |
| Pd-PPh2-MCM-41 | CuBr | H | 94 | 98 | 92 |
| Pd(PPh2)3Cl2 | CuI | H | 97 | 99 | 96 |
Comparing the catalytic performances between Pd-PPh2-MSN-2 and Pd-PPh2-MCM-41 with similar Pd loadings, it was obvious that the former was more active and selective, especially in water-medium Sonogashira reactions. This could be mainly attributed to the uniform nanospheres with shorter pore channels. According to the adsorption test in Fig. 6, Pd-PPh2-MSN-2 exhibited a higher saturated adsorption capacity than Pd-PPh2-MCM-41, which could be attributed to higher SBET. Meanwhile, Pd-PPh2-MSN-2 also showed a faster adsorption rate than Pd-PPh2-MCM-41, implying that the short pore channels might facilitate the diffusion of RhB molecules. Thus, the higher activity of Pd-PPh2-MSN-2 when compared to that of Pd-PPh2-MCM-41 can be attributed to both the higher adsorption of reactant molecules and the lower diffusion limit. The higher selectivity of Pd-PPh2-MSN-2 when compared to that of Pd-PPh2-MCM-41 can be explained according to the reaction mechanism of Sonogashira reactions (Scheme S3, ESI†), which usually forms two products.14 The target product diphenylacetylene could be attributed to the cross-coupling reaction between phenylacetylene and iodobenzene and the byproduct 1,4-diphenylbutadiyene to the self-coupling reaction of phenylacetylene. The coupling of the phenylacetylene intermediate, resulted from activation by Cu(I) in CuI, with the iodobenzene intermediate, generated from activation by Pd(II), plays a key role in determining the selectivity. Obviously, Pd-PPh2-MSN-2 was favorable for the contact and combination of these two intermediates owing to easy diffusion inside short mesopore channels, leading to high selectivity for diphenylacetylene. However, the long distance diffusion in mesoporous channels of the Pd-PPh2-MCM-41 catalyst might supply more opportunities for the self-coupling reaction of phenylacetylene since the phenylacetylene intermediates could not contact iodobenzene intermediates sufficiently, leading to decreased selectivity for the target product. In order to prove the above hypothesis, CuI was replaced by the CuBr with low activity for activating phenylacetylene.14 As shown in Table 1, the selectivity to the target product increased from 90% to 98% since the phenylacetylene intermediates produced slowly on the CuI which supplied more opportunities for their combination with the iodobenzene intermediates. Further evidence was that, despite the higher activity, Pd-PPh2-MSN-2 exhibited almost the same selectivity (98%) as Pd-PPh2-MCM-41 in the water-medium Ullmann reaction with iodobenzene as the single reactant (see Table 3) since only iodobenzene was activated by Pd(II) and no cross-coupling reactions occurred in this system.
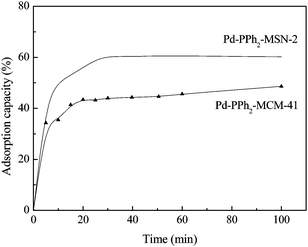 | ||
| Fig. 6 Adsorption test of the Pd-PPh2-MSN-2 and Pd-PPh2-MCM-41 catalysts. Adsorption conditions: 0.050 g catalyst, 250 ml aqueous solution containing 20 mg L−1RhB, T = 25 °C. | ||
Fig. 7 revealed that the Pd-PPh2-MSN-2 could be used repeatedly at least 6 times in water-medium Barbier reactions, showing superiority over the corresponding Pd(PPh3)2Cl2 homogeneous catalyst for reducing cost and diminishing environmental pollution from heavy metal ions. A remarkable decrease in activity (conversion) was observed after 6 cycles, but the selectivity still remained almost unchanged. The ICP analysis demonstrated that only about 3.5% of the Pd species leached off after 6 cycles, suggesting that the loss of Pd active sites can be neglected. The decrease in the activity thus can be mainly attributed to the damage of the catalyst structure. As shown in Fig. 8, the N2 adsorption-desorption isotherm, the low-angle XRD pattern, and the TEM image clearly demonstrate that the damage of the ordered mesoporous structure in the Pd-PPh2-MSN-2 catalyst after being used repeatedly 6 times, leading to an abrupt decrease in SBET, VP and DP (see Table 1).
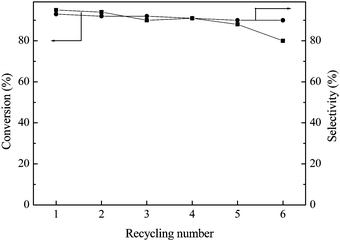 | ||
| Fig. 7 The recycling test of the Pd-PPh2-MSN-2 catalyst in the water-medium Barbier reaction with benzaldehyde and allyl bromide as reactants. Reaction conditions are given in Table 1. | ||
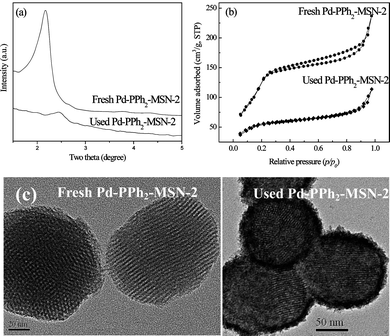 | ||
| Fig. 8 (a) Low-angle XRD patterns, (b) N2 adsorption-desorption isotherms and (c) TEM images of fresh Pd-PPh2-MSN-2 and the Pd-PPh2-MSN-2 catalyst after being used repeatedly 6 times. | ||
Conclusions
This work supplies a new approach to prepare a Pd(II) organometallic catalyst covalently bonded to uniform silica nanospheres with ordered mesoporous channels. In comparison with those bonded to traditional mesoporous silica supports (e.g., MCM-41), the present catalyst exhibits higher activity and even better selectivity in water-medium C–C coupling reactions, which can be attributed to easy diffusion inside the short pore channels. This catalyst displayed comparable catalytic efficiency with the traditional homogeneous catalyst, Pd(PPh3)2Cl2 and could be used repeatedly, showing good potential for industrial applications. Other organometallic catalysts including Rh(I), Ru(II), Ni(II), and rare earth metals could also be designed in this way, which may offer more opportunities for developing powerful and reusable catalysts for green organic syntheses.Acknowledgements
This work was supported by the National Natural Science Foundation of China (20825724) and Shanghai Local Government (10dj1400100, 10PJ1408200, S30406, 07dz22303).Note and references
- (a) U. M. Lindstrom, Chem. Rev., 2002, 102, 2751 CrossRef; (b) C. J. Li, Chem. Rev., 2005, 105, 3095 CrossRef CAS.
- Y. Hayashi, Angew. Chem., Int. Ed., 2006, 45, 8103 CrossRef CAS.
- C. J. Li, Chem. Rev., 1993, 93, 2023 CrossRef CAS.
- I. T. HorváthPaul and T. Anastas, Chem. Rev., 2007, 107, 2169 CrossRef CAS.
- D. A. Ruddy and T. D. Tilley, J. Am. Chem. Soc., 2008, 130, 11088 CrossRef CAS.
- Y. M. A. Yamada, K. Takeda, H. Takahashi and S. Ikegami, J. Org. Chem., 2003, 68, 7733 CrossRef CAS.
- H. X. Li, H. Yin, F. Zhang, H. Li, Y. N. Huo and Y. F. Lu, Environ. Sci. Technol., 2009, 43, 188 CrossRef CAS.
- (a) H. X. Li, F. Zhang, H. Yin, Y. Wan and Y. F. Lu, Green Chem., 2007, 9, 500 RSC; (b) H. X. Li, W. Chai, F. Zhang and J. Chen, Green Chem., 2007, 9, 1223 RSC; (c) H. X. Li, J. Chen, Y. Wan, W. Chai, F. Zhang and Y. F. Lu, Green Chem., 2007, 9, 273 RSC.
- J. L. Gu, W. Fan, A. Shimojima and T. Okubo, Small, 2007, 3, 1740 CrossRef CAS.
- F. Zhang, G. H. Liu, H. X. Li and Y. F. Lu, Adv. Funct. Mater., 2008, 18, 1.
- H. X. Li, F. Zhang, Y. Wan and Y. F. Lu, J. Phys. Chem. B, 2006, 110, 22942 CrossRef CAS.
- J. L. Huang, F. X. Zhu, W. H. He, F. Zhang, W. Wang and H. X. Li, J. Am. Chem. Soc., 2010, 132, 1492 CrossRef CAS.
- B. M. Choudary, K. R. Kumar, Z. Jarnil and G. Thyagarajan, J. Chem. Soc., Chem. Commun., 1985, 931 RSC.
- D. Y. Zhao, Q. Huo, J. Feng, B. F. Chmelka and G. D. Stucky, J. Am. Chem. Soc., 1998, 120, 6024 CrossRef CAS.
- L. Kurti, B. Czako, Strategic Applications of Named Reactions in Organic Synthesis, Science Press, Beijing, 2007, pp. 424 Search PubMed.
- R. A. Sheldon, M. I. Wallau, W. C. E. Arends and U. Schuchardt, Acc. Chem. Res., 1998, 31, 485 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: TG/DTA curves, 31P NMR spectra, FESEM images, Nitrogen adsorption-desorption isotherms, and pore size distribution curves are available free of charge via the Internet. See DOI: 10.1039/c0sc00652a |
| This journal is © The Royal Society of Chemistry 2011 |