Hydroxyl-directed C–H carbonylation enabled by mono-N-protected amino acid ligands: An expedient route to 1-isochromanones†
Yi
Lu
,
Dasheng
Leow
,
Xisheng
Wang
,
Keary M.
Engle
and
Jin-Quan
Yu
*
Department of Chemistry, The Scripps Research Institute (TSRI), 10550 N. Torrey Pines Rd., La Jolla, CA 92037, USA. E-mail: yu200@scripps.edu; Fax: (+1) 858-784-2409; Tel: (+1) 858-784-2433
First published on 18th March 2011
Abstract
A Pd(II)-catalyzed C–H carbonylation protocol of phenethyl alcohols has been developed using amino acid ligands to promote the reaction. This transformation provides an expedient route to 1-isochromanone motifs, which are common structural elements in natural products and other biologically active compounds. A concise synthesis of a histamine release inhibitor showcases the utility of this transformation.
Introduction
During the past decades, palladium-catalyzed C–H functionalization has received a tremendous amount of attention from the chemical community.1,2 Nevertheless, truly practical transformations of this type remain few in number. One of the pervasive challenges in this area is designing reactions that are compatible with common organic substrates where the inherent functionality of the molecule can be used to chelate the metal and facilitate selective C–H cleavage.3 Using ubiquitous chemical functionality (e.g., carboxylic acids and amines) to direct C–H functionalization is advantageous because the directing group can be naturally incorporated into the final product. Aromatic and aliphatic alcohols are a major class of molecules that map directly onto numerous natural products and other biologically active small molecules; thus, they are highly attractive substrates for C–H functionalization. However, metal-catalyzed hydroxyl-directed C–H functionalization methods remain rare.4,5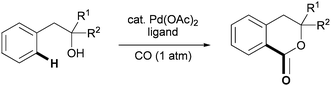 | (1) |
As part of our interest in developing versatile new palladium-catalyzed C–H functionalization reactions to assemble heterocyclic ring systems,6 herein, we report a novel Pd(II)-catalyzed C–H carbonylation protocol for phenethyl alcohols (eqn (1)). This method represents the first example of lactone formation using palladium-catalyzed C–H carbonylation,7 allowing for the rapid preparation of 1-isochromanones, which are key structural motifs in natural products and drug candidates. Importantly, this represents a rare case in which, the directing group (which typically needs to be removed after C–H functionalization) and the coupling partner are fully incorporated into the target molecule without further synthetic manipulations.8 We describe a concise synthesis of a histamine release inhibitor (1) via C–H carbonylation that demonstrates the power of this approach.
Carbonylation reactions have proven to be powerful tools in organic synthesis.9 Prominent among them, Pd(0)-catalyzed carbonylation of aryl and vinyl halides was first reported by Heck and co-workers in 1974,10 and has since become increasingly useful with the discovery of new ligands, nucleophiles, and activating groups (e.g., tosylates and mesylates).11 However, depending on the context, regioselective introduction of the halide is not always straightforward, sometimes requiring several additional steps. It is therefore desirable to develop C–H carbonylation reactions as a complementary approach.
Pd(II)-catalyzed C–H carbonylation of aromatic solvents was first observed by Fujiwara and co-workers in 1980.12,13 However, the synthetic utility of this transformation was limited because the arene substrates needed to be used in excess (typically as the solvent) and because intractable mixtures of regioisomeric products were obtained when substituted arenes were used. Orito and co-workers overcame these problems through the use of an amine directing group,7 which enhanced the reactivity and allowed for precise control of the regioselectivity. This C–H carbonylation reaction led to efficient formation of the corresponding benzolactam products. In 2008, our group developed a carboxylate directed C–H carbonylation reaction of benzoic and phenylacetic acids with CO,14 and subsequently, arene carbonylation has also been reported using other directing groups.15–18
With these precedents in mind, we became interested in developing new methods to synthesize different carbonyl-containing motifs that are prevalent in bioactive molecules, with the long-term aim of devising technology to prepare biologically significant ring structures in a modicum of steps from simple starting materials. To this end, we identified 1-isochromanones as appealing targets and hypothesized that they could arise from a hydroxyl-directed ortho-C(sp2)–H carbonylation reaction of phenethyl alcohols (eqn (1)).
1-Isochromanones are six-membered cyclic lactones that are commonly found in biologically active small molecules (Fig. 1). For example, they are prevalent in mycotoxins, a family of natural products that are secondary metabolites produced by toxigenic fungi.19aMycotoxins are a cause for concern in food safety, and they display a range of different biological activity. For example, dehydroaltenusin has been shown to be a specific inhibitor of mammalian DNA polymeraseα.19b,c Intriguing bioactivity has been found with other 1-isochromanones derivatives as well. For example, one class of compounds was found to be potent urotensin-II receptor agonists.20a Spiro-isochroman-piperidine compounds were found to possess analgesic activity in inhibiting the induced release of histamines from isolated rat peritoneal mast cells.20b 1-Isochromanones are traditionally synthesized viaPd(0)-catalyzed carbonylation of aryl halides21 or directed ortho-lithiation.22
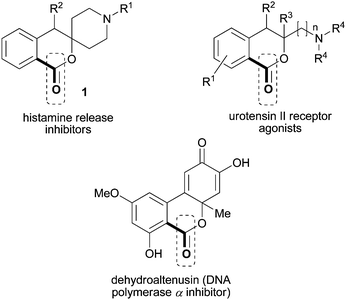 | ||
| Fig. 1 Biologically active 1-isochromanones: lead compounds and natural products. | ||
Our C–H functionalization approach for synthesizing 1-isochromanones relies upon hydroxyl-directed Pd(II)-mediated ortho-C–H cleavage and subsequent CO insertion into the Pd–C bond. Our group recently demonstrated that hydroxyl groups could efficiently direct ortho-C–H cleavage, a mode of reactivity that we exploited to develop novel Pd(II)-catalyzed C–H olefination and intramolecular C–O cyclization reactions.4 There are several issues to consider when using a hydroxyl directing group in C–H functionalization:4,21,23 (1) benzylic alcohols can undergo β-carbon elimination; (2) coordination between Pd(II) and the hydroxyl group is weak under neutral conditions; and (3) primary or secondary alcohols can undergo oxidation or β–hydride elimination during the reaction to give ketones or aldehydes. Moreover, there are unique challenges in developing a Pd(II)-catalyzed C–H carbonylation protocol: (1) excess CO will coordinatively saturate the Pd(II) metal center, thereby preventing formation of the requisite C–H agostic interaction; and (2) under a CO environment, Pd(II) usually reduces to catalytically inactive Pd(0).24
Results and discussion
Initial studies and reaction optimization
In our initial screening studies, we selected tertiary alcohol 2a in order to avoid unwanted alcohol oxidation as we tested different reaction conditions (Table 1). Our first attempts were unsuccessful, as none of the desired product was observed when we tried to perform C–H carbonylation of phenethyl alcohol 2a using previously developed conditions.14 After a brief survey of alternative conditions, we found that the C–H carbonylation proceeded in 12% conversion in the presence of Pd(OAc)2 (20 mol%), Li2CO3 (1.0 equiv.), and dichloromethane (DCM) under CO (1 atm) at 80 °C for 48 h (see Supporting Information†). Through extensive optimization, we found that AgOAc was an optimal oxidant for the reaction (see Supporting Information†). However, the conversion did not improve beyond 35%, presumably due to the decomposition of the catalyst to palladium black under the reductive CO atmosphere.
| Entry | Ligand | Conv. (%) | Entry | Ligand | Conv. (%) |
|---|---|---|---|---|---|
| a The reaction conversion was determined by 1H NMR analysis of the crude reaction mixture using CH2Br2 as the internal standard. b 110 °C. Boc = tert-butyloxycarbonyl, Ac = acetyl, Fmoc = fluorenylmethyloxycarbonyl, Bz = benzoyl, (+)-Men = (+)-menthyl(O2C), Ile = L-isoleucine, Ala = L-alanine, Phe = L-phenylalanine, Ser = L-serine, Nva = L-norvaline, Nle = L-norleucine, Acpc = 1-aminocyclopropane-1-carboxylic acid, Leu = L-leucine. | |||||
| 1 | — | 35 | 9 | Boc-Acpc-OH | 53 |
| 2 | Boc-Ile-OH · 0.5 H2O | 46 | 10 | Boc-tert-Leu-OH | 7 |
| 3 | Boc-Ala-OH | 27 | 11 | Boc-Leu-OH · H2O | 66 |
| 4 | Boc-Phe-OH | 26 | 12 | Ac-Leu-OH | 35 |
| 5 | Boc-Ser-OH | 5 | 13 | Fmoc-Leu-OH | 20 |
| 6 | Boc-Nva-OH | 54 | 14 | Bz-Leu-OH | 12 |
| 7 | Boc-Nle-OH | 34 | 15 | (+)-Men-Leu-OH | 67 |
| 8 | Boc-α-Me-Ala-OH | 5 | 16 | (+)-Men-Leu-OH | 84b |
Based on our group's recent discovery that mono-N-protected amino acids are capable of accelerating Pd(II)-catalyzed C–H olefination reactions,25 we turned our attention to this class of ligands, hypothesizing that they could reduce the rate of Pd(II) reduction to palladium black in the presence of CO. Thus, we extensively surveyed a variety of amino acids ligands. Among the N-Boc-protected amino acids that were tested, Boc-Leu-OH·H2O increased the yield to 66% (Table 1, entry 11). Several N-protecting groups such as Ac, Fmoc, Bz and (+)-menthyl(O2C) were examined (Table 1, entries 12–15). Ac-Leu-OH, Fmoc-Leu-OH and Bz-Leu-OH all gave poor conversions, while (+)-menthyl(O2C)-Leu-OH and Boc-Leu-OH·H2O gave similar conversions. We assessed the effect of reaction temperature on the conversion and found that, by increasing the temperature from 80 °C to 110 °C, the conversion was further improved to 84% (Table 1, entry 16)
Substrate scope
With these optimized conditions in hand, we then examined the substrate scope for this reaction.‡ In general, substrates bearing electron-rich aryl groups (Table 2, entries 3b–d, 3f–h) gave the best yields. Electron-withdrawing groups such as fluorides, chlorides and bromides were tolerated on the aromatic ring (Table 2, entries 3i–o) but led to lower yields as compared to the electron-rich aryl substrates. It is worth noting that under these reaction conditions, the C–Br bond remained intact (Table 2, entry 3n), which could not be achieved by Pd(0) catalysis.21 Stronger electron-withdrawing substituents gave substantially lower product yields. This observation serves as qualitative evidence that the C–H cleavage step might proceed via electrophilic palladation.26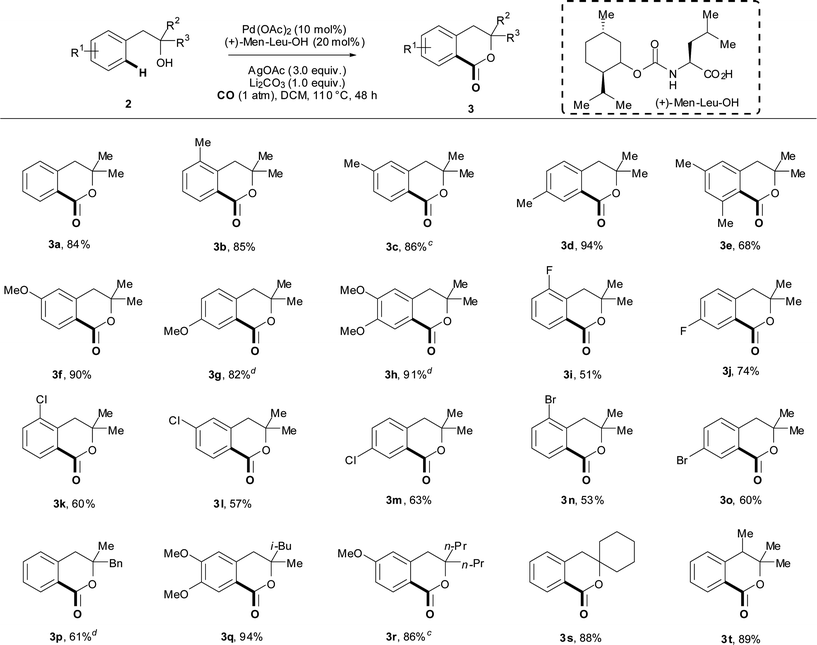
Phenethyl alcohols with substituents at the meta position reacted exclusively at the less sterically hindered position, leading to formation of single regioisomers (Table 2, entries 3c, 3f, 3l, 3r). We examined one 3,5-disubstituted substrate, in which both ortho-C–H derivatives were sterically encumbered, and observed a slightly diminished yield (Table 2, entry 3e). Finally, we probed the effect of various substituents on the alkyl chain adjacent to the hydroxyl directing group. Different levels of steric bulk were not found to diminish the yield (Table 2, entries 3p–r). The presence of a cyclohexyl ring led to the formation of a spirocyclic product in excellent yield (Table 2, entry 3s), which paved the way for the synthesis of biologically active molecules containing this structural motif. Substitution at the benzylic position was also tolerated (Table 2, entry 3t). When we exposed primary and secondary phenethyl alcohols to these reaction conditions, we predominantly observed oxidative decomposition along with less than 50% yield of the desired product (as quantified by 1H NMR using CH2Br2 as an internal standard).
Next, we sought to test the utility of our Pd(II)-catalyzed C–H carbonylation reaction in the synthesis of known bioactive molecules. One of the challenges of using this reaction in this context is the presence of competitively coordinating functional groups that could potentially interfere with the C–H carbonylation process. As a representative example, we elected to target compound 3u, the N-protected version of a histamine release inhibitor (Fig. 2).20b The corresponding starting material, spiro-isochroman-piperidine derivative 2u, could be obtained from commercially available 4-benzyl-4-hydroxypiperidine in one step via routine N-Boc protection. We were delighted to find that phenethyl alcohol 2u reacted smoothly with CO to give the corresponding cyclic lactone 3u in 62% isolated yield. This methodology could potentially be used in drug discovery for the rapid synthesis of an entire library of histamine release inhibitors.
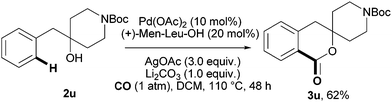 | ||
| Fig. 2 Expedient synthesis of a histamine release inhibitor (3u). | ||
Proposed mechanism
A plausible mechanism for this catalytic transformation is depicted in Fig. 3. The hydroxyl group directs ortho-C–H bond cleavage by Pd(II). Previously, we found that C–H functionalization of phenethyl alcohols could take place in the absence of the base.4 Thus, in accordance with that observation, we propose that the hydroxyl moiety coordinates with Pd(II) as a weak neutral donor. A molecule of CO then binds to the putative [Pd(II)–Ar] species. 1,1-Migratory insertion of CO into the Pd–C bond then takes place to forge the new C–C bond, generating a seven-membered cyclic intermediate. This is followed by nucleophilic attack of the hydroxyl group on the carbonyl moiety to give the final product.7 However, it is also possible that CO actually inserts into the Pd–OR bond. Theoretical studies have shown that under certain conditions, CO preferentially undergoes migratory insertion with Pd–OR bonds.27 Further studies are needed to disambiguate the mechanistic details of this reaction.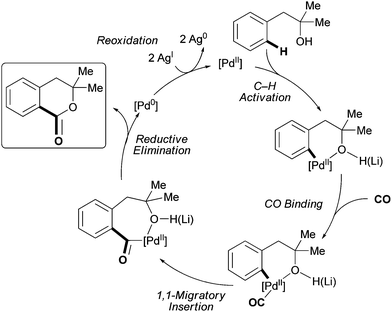 | ||
| Fig. 3 Plausible mechanism for hydroxyl-directed Pd(II)-catalyzed C–H carbonylation. | ||
Conclusions
In conclusion, we have developed a Pd(II)-catalyzed ortho-C–H carbonylation reaction with phenethyl alcohol derivatives enabled by mono-N-protected amino acid ligands. This represents a rare example of hydroxyl-directed C–H functionalization. Our new protocol allows for one-step access to 1-isochromanones, which are common structural motifs in natural products and pharmaceutical agents. This approach obviates the need for pre-functionalization to generate aryl halide starting materials and utilizes carbon monoxide, which is an abundant C1carbon feedstock source. Importantly, in this reaction, the phenethyl alcohol substrate and the coupling partner, CO, are both fully incorporated into the desired product. As such, this method represents an encouraging step forward towards improving atom-28 and step-economy29 in organic synthesis. Additionally, the current investigation further establishes the breadth and general applicability of amino acid ligand–promoted C–H functionalization. Finally, we demonstrate the utility of C–H carbonylation by synthesizing structurally intricate histamine release inhibitors from simple starting materials, which showcases how molecular complexity can be drastically advanced in a single step using C–H functionalization. Efforts towards an asymmetric variant of this transformation and a hydroxyl-directed C(sp3)–H carbonylation protocol are currently underway in our laboratory and will be reported in due course.Acknowledgements
We gratefully acknowledge TSRI and the US NSF (NSF CHE-1011898) for financial support. Y. L. is a China Scholarship Council Exchange Scholar from the State Key Laboratory of Coordination Chemistry, School of Chemistry and Chemical Engineering, Nanjing University. D. L. thanks A*STAR for a postdoctoral fellowship. K. M. E. thanks the US NSF, US DOD, and the Skaggs-Oxford Scholarship Program for predoctoral fellowships.Notes and references
- For book chapters on transition metal–catalyzed C–H functionalization, see: (a) Topics in Current Chemistry, ed. J.-Q. Yu and Z. Shi, Springer-Verlag, Berlin, 2010, vol. 292 Search PubMed; (b) J. Hartwig, Organotransition Metal Chemistry: From Bonding to Catalysis, University Science Books, Sausalito, 2010, pp. 825–875 Search PubMed.
- For selected reviews of palladium-catalyzed C–H functionalization, see: (a) C. Jia, T. Kitamura and Y. Fujiwara, Acc. Chem. Res., 2001, 34, 633–639 CrossRef CAS; (b) V. Ritleng, C. Sirlin and M. Pfeffer, Chem. Rev., 2002, 102, 1731–1769 CrossRef CAS; (c) D. Alberico, M. E. Scott and M. Lautens, Chem. Rev., 2007, 107, 174–238 CrossRef CAS; (d) I. V. Seregin and V. Gevorgyan, Chem. Soc. Rev., 2007, 36, 1173–1193 RSC; (e) B.-J. Li, S.-D. Yang and Z. Shi, Synlett., 2008, 949–957 CAS; (f) O. Daugulis, H.-Q. Do and D. Shabashov, Acc. Chem. Res., 2009, 42, 1074–1086 CrossRef CAS; (g) L. Ackermann, R. Vicente and A. R. Kapdi, Angew. Chem., Int. Ed., 2009, 48, 9792–9826 CrossRef CAS; (h) T. W. Lyons and M. S. Sanford, Chem. Rev., 2010, 110, 1147–1169 CrossRef CAS; (i) R. Jazzar, J. Hitce, A. Renaudat, J. Sofack-Kreutzer and O. Baudoin, Chem.–Eur. J., 2010, 16, 2654–2672 CrossRef CAS.
- (a) J.-Q. Yu, R. Giri and X. Chen, Org. Biomol. Chem., 2006, 4, 4041–4047 RSC; (b) R. Giri, B.-F. Shi, K. M. Engle, N. Maugel and J.-Q. Yu, Chem. Soc. Rev., 2009, 38, 3242–3272 RSC; (c) X. Chen, K. M. Engle, D.-H. Wang and J.-Q. Yu, Angew. Chem., Int. Ed., 2009, 48, 5094–5115 CrossRef CAS.
- (a) Y. Lu, D.-H. Wang, K. M. Engle and J.-Q. Yu, J. Am. Chem. Soc., 2010, 132, 5916–5921 CrossRef CAS; (b) X. Wang, Y. Lu, H.-X. Dai and J.-Q. Yu, J. Am. Chem. Soc., 2010, 132, 12203–12205 CrossRef CAS.
- E. M. Simmons and J. F. Hartwig, J. Am. Chem. Soc., 2010, 132, 17092–17095 CrossRef CAS.
- For representative examples from our group, see: (a) J.-J. Li, T.-S. Mei and J.-Q. Yu, Angew. Chem., Int. Ed., 2008, 47, 6452–6455 CrossRef CAS; (b) M. Wasa and J.-Q. Yu, J. Am. Chem. Soc., 2008, 130, 14058–14059 CrossRef CAS.
- For a pioneering example of Pd(II)-catalyzed lactam-forming C–H carbonylation, see: K. Orito, A. Horibata, T. Nakamura, H. Ushito, H. Nagasaki, M. Yuguchi, S. Yamashita and M. Tokuda, J. Am. Chem. Soc., 2004, 126, 14342–14343 Search PubMed.
- Discounting the formal loss of H2, every atom of the starting material and the coupling partner is directly incorporated into the product. A related class of reactions is Pd(II)-catalyzed oxidative C–H/C–H cross-coupling; for recent examples of biaryl formation using this approach, see: (a) R. Li, L. Jiang and W. Lu, Organometallics, 2006, 25, 5973–5975 CrossRef CAS; (b) J.-B. Xia and S.-L. You, Organometallics, 2007, 26, 4869–4871 CrossRef CAS; (c) D. R. Stuart and K. Fagnou, Science, 2007, 316, 1172–1175 CrossRef CAS; (d) T. A. Dwight, N. R. Rue, D. Charyk, R. Josselyn and B. DeBoef, Org. Lett., 2007, 9, 3137–3139 CrossRef CAS; (e) K. L. Hull and M. S. Sanford, J. Am. Chem. Soc., 2007, 129, 11904–11905 CrossRef CAS; (f) B.-J. Li, S.-L. Tian, Z. Fang and Z.-J. Shi, Angew. Chem., Int. Ed., 2008, 47, 1115–1118 CrossRef CAS; (g) G. Brasche, J. García-Fortanet and S. L. Buchwald, Org. Lett., 2008, 10, 2207–2210 CrossRef CAS; (h) X. Zhao, C. S. Yeung and V. M. Dong, J. Am. Chem. Soc., 2010, 132, 5837–5844 CrossRef CAS.
- For reviews on Pd(0)-catalyzed carbonylation reactions of aryl halides, see: (a) Handbook of Organopalladium Chemistry for Organic Synthesis, ed. E. Negishi and A. de Meijere, John Wiley & Sons, Inc., New York, 2002, vol. 2, pp. 2309–2741 Search PubMed; (b) A. Brennführer, H. Neumann and M. Beller, Angew. Chem., Int. Ed., 2009, 48, 4114–4133 CrossRef.
- (a) A. Schoenberg, I. Bartoleti and R. F. Heck, J. Org. Chem., 1974, 39, 3318–3326 CrossRef CAS; (b) A. Schoenberg and R. F. Heck, J. Org. Chem., 1974, 39, 3327–3331 CrossRef CAS.
- R. H. Munday, J. R. Martinelli and S. L. Buchwald, J. Am. Chem. Soc., 2008, 130, 2754–2755 CrossRef CAS.
- Y. Fujiwara, T. Kawauchi and H. Taniguchi, J. Chem. Soc., Chem. Commun., 1980, 220–221 RSC.
- For examples of C(sp2)–H alkoxycarbonylation of palladacycles, see: (a) P. S. Pregosin and R. Rüedi, J. Organomet. Chem., 1984, 273, 401–413 CrossRef CAS; (b) G. Balavoine, J. C. Clinet, P. Zerbib and K. Boubekeur, J. Organomet. Chem., 1990, 389, 259–275 CrossRef CAS.
- R. Giri and J.-Q. Yu, J. Am. Chem. Soc., 2008, 130, 14082–14083 CrossRef CAS.
- C. Houlden, M. Hutchby, C. Bailey, J. Ford, S. Tyler, M. Gagné, G. Lloyd-Jones and K. Booker-Milburn, Angew. Chem., Int. Ed., 2009, 48, 1830–1833 CrossRef CAS.
- R. Giri, J. K. Lam and J.-Q. Yu, J. Am. Chem. Soc., 2010, 132, 686–693 CrossRef CAS.
- B. Haffemayer, M. Gulias and M. J. Gaunt, Chem. Sci., 2011, 2, 312–315 RSC.
- For an intriguing ortho-ethoxycarbonylation of 2-phenylpyridine substrates using diethyl azodicarboxylate as the carboxyl donor, see: W.-Y. Yu, W. N. Sit, K.-M. Lai, Z. Zhou and A. S. C. Chan, J. Am. Chem. Soc., 2008, 130, 3304–3306 Search PubMed.
- For a review of mycotoxins, see: (a) S. Bräse, A. Encinas, J. Keck and C. F. Nising, Chem. Rev., 2009, 109, 3903–3990 CrossRef. For isolation and activity screenings of dehydroaltenusin, see: (b) T. Rosett, R. H. Sankhala, C. E. Stickings, M. E. U. Taylor and R. Thomas, Biochem. J., 1957, 67, 390–400 CAS; (c) S. Kamisuki, C. Murakami, K. Ohta, H. Yoshida, F. Sugawara, K. Sakaguchi and Y. Mizushina, Biochem. Pharmacol., 2002, 63, 421–427 CrossRef CAS.
- (a) F. Lehmann, A. Pettersen, E. A. Currier, V. Sherbukhin, R. Olsson, U. Hacksell and K. Luthman, J. Med. Chem., 2006, 49, 2232–2240 CrossRef CAS; (b) M. Yamato, K. Hashigaki, M. Ikeda, H. Ohtake and K. Tasaka, J. Med. Chem., 1981, 24, 194–198 CrossRef CAS.
- (a) A. Cowell and J. K. Stille, J. Am. Chem. Soc., 1980, 102, 4193–4198 CrossRef CAS; (b) W. E. Lindsell, D. D. Palmer, P. N. Preston and G. M. Rosair, Organometallics, 2005, 24, 1119–1133 CrossRef CAS.
- For reviews of directed ortho-lithiation, see: (a) P. Beak and V. Snieckus, Acc. Chem. Res., 1982, 15, 306–312 CrossRef CAS; (b) M. Schlosser and F. Mongin, Chem. Soc. Rev., 2007, 36, 1161–1172 RSC.
- For Pd(0)-catalyzed phenol-directed C–H arylation, see: (a) D. D. Hennings, S. Iwasa and V. H. Rawal, J. Org. Chem., 1997, 62, 2–3 CrossRef CAS; (b) T. Satoh, Y. Kawamura, M. Miura and M. Nomura, Angew. Chem., Int. Ed., 1997, 36, 1740–1742 CrossRef CAS. For an early attempt at Pd(0)-catalyzed ortho-C–H arylation of benzylic alcohols, see: (c) Y. Terao, H. Wakui, T. Satoh, M. Miura and M. Nomura, J. Am. Chem. Soc., 2001, 123, 10407–10408 CrossRef CAS. For Pd(II)-catalyzed phenol-directed C–H olefination, see: (d) M. Miura, T. Tsuda, T. Satoh and M. Nomura, Chem. Lett., 1997, 26, 1103–1104 CrossRef.
- I. I. Moiseev, Pure Appl. Chem., 1989, 61, 1755–1762 CAS.
- (a) D.-H. Wang, K. M. Engle, B.-F. Shi and J.-Q. Yu, Science, 2010, 327, 315–319 CrossRef CAS; (b) K. M. Engle, D.-H. Wang and J.-Q. Yu, Angew. Chem., Int. Ed., 2010, 49, 6169–6173 CrossRef CAS; (c) K. M. Engle, D.-H. Wang and J.-Q. Yu, J. Am. Chem. Soc., 2010, 132, 14137–14151 CrossRef CAS.
- B. Martín-Matute, C. Mateo, D. J. Cárdenas and A. M. Echavarren, Chem.–Eur. J., 2001, 7, 2341–2348 CrossRef CAS.
- S. A. Macgregor and G. W. Neave, Organometallics, 2003, 22, 4547–4556 CrossRef CAS.
- For the concept of atom-economy, see: (a) B. M. Trost, Science, 1991, 254, 1471–1477 CrossRef CAS; (b) B. M. Trost, Angew. Chem., Int. Ed., 1995, 34, 259–281 CrossRef CAS. For a discussion of C–H functionalization as green chemistry, see: (c) C.-J. Li and B. M. Trost, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 13197–13202 CrossRef CAS.
- For the concept of step-economy, see: (a) P. A. Wender, V. A. Verma, T. J. Paxton and T. H. Pillow, Acc. Chem. Res., 2008, 41, 40–49 CrossRef CAS; (b) P. A. Wender and B. L. Miller, Nature, 2009, 460, 197–201 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Data for new compounds and experimental procedures. See DOI: 10.1039/c0sc00633e |
| ‡ General procedure for Pd(II)-catalyzed hydroxyl-directed C–H carbonylation: A 50 mL Schlenk-type sealed tube (with a Teflon high pressure valve and side arm) equipped with a magnetic stir bar was charged with the phenethyl alcohol (2) (0.20 mmol), Pd(OAc)2 (4.5 mg, 0.02 mmol), Li2CO3 (73.8 mg, 0.20 mmol), (+)-menthyl(O2C)-Leu-OH (12.5 mg, 0.04 mmol), AgOAc (100.1 mg, 0.60 mmol), and DCM (5.0 mL). The reaction tube was repeatedly purged and refilled (×3) with CO (1 atm, balloon). The reaction mixture was capped and stirred at 110 °C for 48 h. After being allowed to cool to room temperature, the solution was filtered through a short pad of Celite. The filtrate was concentrated in vacuo and purified by silica gel flash column chromatography using hexanes/EtOAc as the eluent to give the cyclic lactone product (3). |
| This journal is © The Royal Society of Chemistry 2011 |