The programming role of trans-acting enoyl reductases during the biosynthesis of highly reduced fungal polyketides†
Mary N. Heneghan*a, Ahmed A. Yakasaib, Katherine Williamsa, Khomaizon A. Kadira, Zahida Wasilb, Walid Bakeerb, Katja M. Fischb, Andrew M. Baileya, Thomas J. Simpsonb, Russell J. Cox*b and Colin M. Lazarus*a
aSchool of Biological Sciences, University of Bristol, Woodland Road, Bristol, BS8 1UG, UK. E-mail: C.M.Lazarus@bristol.ac.uk
bSchool of Chemistry, University of Bristol, Cantock's Close, Bristol, BS8 1TS, UK. E-mail: r.j.cox@bris.ac.uk; Fax: +44 (0) 117 925 1295; Tel: +44 (0) 117 928 9184
First published on 2nd March 2011
Abstract
A novel polyketide synthase nonribosomal peptide synthetase (PKS-NRPS) genecluster was isolated from Beauveria bassiania 992.05. The cluster encodes the enzymes responsible for the biosynthesis of the new 2-pyridone desmethylbassianin (DMB). DMB is structurally related to tenellin from B. bassiana 110.25 but it differs in chain length and degree of methylation. Despite these programming differences the 20 kb DMB biosynthetic genecluster has 90% sequence identity to the tenellingenecluster. Silencing of the PKS-NRPS gene, dmbS, resulted in total loss of DMB production. Co-expression of dmbS in Aspergillus oryzae with its cognate trans-actingenoyl reductase gene, dmbC, produced predesmethylbassianin A, the first isolable precursor in the biosynthetic pathway. Expression of dmbS with the tenellintrans-actingenoyl reductase gene, tenC, also resulted in the production of predesmethylbassianin A. Co-expression of tenS, the tenellin PKS-NRPS, with dmbC produced pretenellin A. These results show that the tenS and dmbS encoded PKS-NRPS contains the programme for polyketide biosynthesis, while the trans-actingERs appear to control the fidelity of the programme. Expression of a hybrid synthetase in which the PKS of the tenellin synthetase was fused to the NRPS from DMBS produced prototenellins A to C, indicating that the NRPS does not act as a selecting gatekeeper to affect the PKS programme.
Introduction
Highly reduced fungal polyketides are a large family of structually complex secondary metabolites with an extensive range of biological activities.1,2 The genes encoding the biosynthesis of these compounds are usually found in a cluster, encoding a highly reducing polyketide synthase (HR-PKS) and several tailoring enzymes.3 Fungal HR-PKS are iterative, meaning that a single set of catalytic sites works repeatedly to generate the product. Thus, during each cycle of chain extension the modification reactions can be different, suggesting that iterative PKSs are programmed. The precise origin of programming in fungal HR-PKS remains obscure. The structural complexity of fungal HRpolyketides is further enhanced by the existence of hybrid HR-PKS nonribosomal peptide synthetases (NRPS) where the HR-PKS module is responsible for the synthesis of the carbon skeleton and the NRPS module programmes the addition of an amino acid usually followed by Dieckmann cyclisation (Scheme 1). Unlike bacterial modular PKS systems where programming is highly predictable, the structures of polyketides derived from fungal HR-PKS cannot be determined from sequence information alone.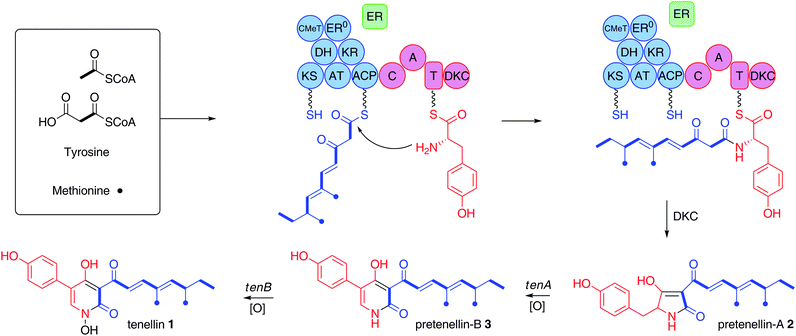 | ||
| Scheme 1 Biosynthesis of tenellin: KS, β-ketoacyl synthase; AT, acyltransferase; DH, dehydratase; ER, enoyl reductase; ER0, defective enoyl reductase; CMeT, C-methyltransferase; KR, β-ketoacylreductase; ACP, acyl carrierprotein; C, condensation; A, adenylation; T, thiolation, DKC, Dieckmann cyclase. Blue, PKS; red, NRPS; green, trans-actingER. | ||
One of the best characterised fungal HR-polyketide pathways is that of the 2-pyridone tenellin 1 from the insect pathogen Beauveria bassiana 110.25 (Scheme 1). The tenellingenecluster isolated from B. bassiana 110.25 consists of four open reading frames comprising tenA and tenB, encoding cytochrome P450 mono-oxygenases; tenC, a trans-actingenoyl reductase; and tenS, which is an approximately 12 kb gene encoding an iterative HR-PKS fused to a single module NRPS (Scheme 1).4 The precise steps of tenellinbiosynthesis have been determined through coordinated heterologous expression of the pathway genes in Aspergillus oryzae and RNAi silencing experiments in the native organism.5,6,7 Coordinated heterologous expression of tenS and tenC leads to the production of pretenellin-A 2 with the correctly elaborated polyketide side chain derived from acetate, malonate and methionine, and where the tetramic acid is constructed by condensation with tyrosine, Dieckmann cyclisation and release (Scheme 1).7,8 Addition of tenA results in the production of pretenellin-B 3, demonstrating that tenA is responsible for oxidative ring expansion,5 while heterologous expression of all four genes produces tenellin1, with tenB catalysing the final N-hydroxylation step.7
Tenellin-like structures which show clear differences in their intrinsic polyketide programming, have been reported from other Beauveria species (Fig. 1). For example bassianin 4, a hexaketide, is reportedly produced by Beauveria tenella and differs from tenellin by one cycle of chain extension.9 Comparison of the geneclusters responsible for the synthesis of structurally related compounds could provide insight and answer key questions about the programming of HR-PKS-NRPS systems.
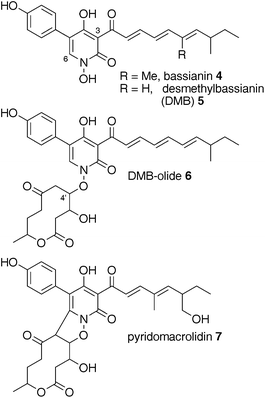 | ||
| Fig. 1 2-Pyridones from Beauveria species. | ||
In order to address this aim we set out to clone a biosynthetic genecluster for one of these tenellin-related compounds to enable us to directly compare the biosynthesis of closely related—but differently programmed—highly reduced polyketides.
Results
Screening of Beauveria strains and isolation of new 2-pyridones
Bassianin 4 was reported over thirty five years ago as a product of B. tenella,9 but we were unable to obtain the original strain. In an attempt to discover an alternative producer thirty Beauveria strains were obtained from public culture collections and grown in liquid media (TPM) known to support 2-pyridone production. Organic extracts from the media and mycelia were prepared and these were examined by analytical LCMS. One strain (992.05) produced new compounds which were identified by LCMS analysis as likely 2-pyridones because of their characteristic UV maxima at 340 nm with odd-valued masses indicating the presence of nitrogen. B. bassiana 992.05 was therefore grown at preparative scale and preparative mass-directed HPLC was performed to purify a sufficient amount of each compound for structure elucidation by NMR.The first compound was isolated by selecting mass ion 382 ([M]H+) for purification. This afforded 7.6 mg (7.6 mg L−1) of a compound with HRMS 404.1472 ([M]Na+) consistent with the molecular formula C22H23NO5Na (calc. 404.1474). The 500 MHz 1H NMR spectrum indicated a methyl triplet and a methyl doublet, an allylic methine multiplet and (as confirmed by COSY) six contiguous olefinic protons. HMBC data linked the terminal olefinic proton to a carbonyl and a typical C-3 quaternary carbon of a 2-pyridone. The presence of the 2-pyridone was further confirmed by the characteristic 6-H singlet. Finally 1H NMR also revealed a p-substituted aromatic nucleus. Complete HSQC and HMBC characterisation and comparison with tenellin confirmed the structure as desmethylbassianin (DMB, 5).
The second, later eluting, compound, was purified by selecting mass ion 580 ([M]H+) for purification. This afforded 6.4 mg (6.4 mg L−1) of a compound with HRMS 580.2540 consistent with the molecular formula C32H38NO9 (calc. 580.2547). 1H NMR at 500 MHz revealed the same spectrum as 5, overlayed with a further methyl doublet, three methine-heteroatom multiplets and possible methylene signals. This indicated that the DMB skeleton was intact with another C10H12O4 moiety attached. COSY indicated that none of the new resonances coupled to any of the DMB resonances. COSY, HSQC and HMBC spectra confirmed that the extra signals emanated from a decanolide, while nOe difference spectra indicated that the decanolide was attached to the pyridonevia the N-hydroxyl at C-4′ (Fig. 1). This compound, 6, was named “DMB-olide” and shows significant similarity to the known compound pyridomacrolidin7 from B. bassiana EPF-5.10
Isolation of the DMBgenecluster
A Southern blot of B. bassiana 992.05 genomic DNA using a tenSprobe indicated that there was only one tenS-like gene within the genome. A PCR-based approach was then adopted to isolate the DMBgenecluster (Fig. 2). The high level of structural similarity between tenellin1 and DMB5, coupled with the very close relationship between the producing organisms, suggested that the genes should be highly similar and we speculated that PCR primers matching the known tengenes might amplify their dmborthologues. Thus dmbA and dmbB, encoding cytochrome P450oxidases, and dmbC, encoding a trans-actingenoyl reductase, were each amplified from B. bassiana 992.05 genomic DNA using tenellin-based primers.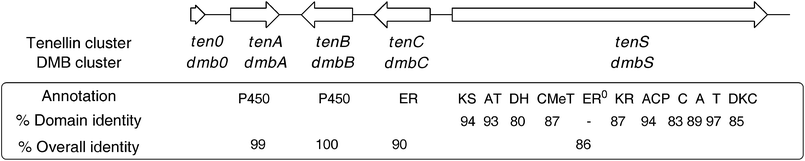 | ||
| Fig. 2 Comparison of the tenS and dmbSgeneclusters. See Scheme 1 for domain abbreviations. Identities at peptide level. | ||
Tenellin primers were then used to amplify 540 bp at the 5′ end of the ketosynthase domain and 472 bp at the 3′ end of the Dieckmann cyclase domain of the putative dmbSgene. A combination of dmbS-specific and tenS primers were then used to amplify the entire dmbS coding region as five overlapping products (P2–P6, see ESI†). To isolate the intergenic fragments, primers were designed based on the orientation of the tenellingenecluster. All four intergenic fragments were cloned. All PCR products were then sequenced and DNA sequences were assembled using Sequencher® analysis software producing a 20,147 bp contig.
Sequence analysis of the DMBgenecluster
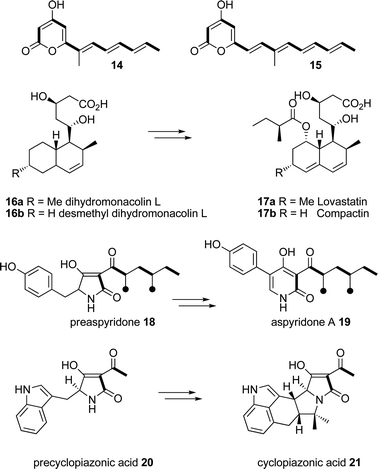
Sequence analysis of the entire contig revealed a genecluster structure identical to that of the tenellincluster, with 1 partial and 4 complete open reading frames (Fig. 2). The entire putative DMBgenecluster has a 90% identity value (ClustalW) to the tenellingenecluster at the nucleotide level. The four complete ORFs are arranged in the same orientation as their tenellin counterparts, and individually have identities of translated peptides of between 86% and 100% with them. They contain the same number of introns, which are located at identical positions within the coding sequence.The dmbS and tenSgenes are homologous in their domain organisation i.e. they encode β-ketoacyl synthase (KS), acyltransferase (AT), dehydratase (DH), C-methyltransferase (CMet), β-ketoacyl-reductase (KR) and acyl carrierprotein (ACP) domains typical of a HR-PKS, followed by condensation (C), adenylation (A), thiolation (T) and Dieckmann cyclase (DKC) domains characteristic of an NRPS module. Like tenS, the dmbS encoded ER0 domain lacks key protein motifs which suggests it is inactive. Detailed domain analysis of the translated PKS-NRPS was carried out (see ESI†) which showed identities of between 80 and 97% (Fig. 2). The consensus sequences for each domain were conserved across both synthetases. No significant differences in sequence could be found which might be linked to the differences in programme between TENS and DMBS.
Inactivation of dmbS
To confirm the involvement of dmbS in the biosynthesis of 5 and 6, inactivation of the PKS-NRPS gene was performed. The preferred method for functional gene analysis in fungi is targeted gene disruption viatransgene integration (gene knockout). However, homologous recombination can be difficult to achieve in filamentous fungi, and when it does occur targeting efficiencies are often low (0.5–10%).11 Post-transcriptional gene silencing (PTGS) methods such as antisense or RNAi have emerged as useful new tools in the suppression of gene expression and have been demonstrated in a variety of fungi.7,12,13 Both targeted gene disruption and antisense-mediated suppression have previously been demonstrated in B. bassiana.5To perform functional analysis on dmbS a dual silencing/knockout approach using a single construct was adopted. Typically knockout vectors are constructed to contain a large section of a gene (∼4 kb) divided into two sections termed the left and right arms (LA, RA). The LA and RA are separated by a selectable marker so that homologous recombination of the LA and RA with the native gene results in insertion of the selectable marker into the gene, disrupting its function. In our dual approach the knockout vector contained an additional heterologous promoter between the LA and the selectable marker (Fig. 3). If ectopic integration occurs (i.e. no gene disruption) the heterologous promoter will drive antisense expression of the LA triggering the PTGS pathway. B. bassiana has been shown to be sensitive to the herbicideglufosinate (basta) and this was therefore selected as the resistance marker for the construct.4,5 The constitutive gpdA promoter from A. nidulans is already known to function in Beauveria species to drive antisense RNA production.
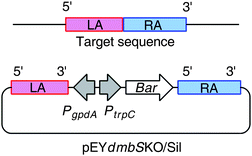 | ||
| Fig. 3 Dual KO/silencing strategy used for investigation of dmbS function: PtrpC - trpC Promoter; PgpdA, gpdA Promoter; Bar, glufosinate resistance structural gene; RA, right arm; LA, left arm. | ||
B. bassiana 992.05 was transformed with this plasmid and 62 basta-resistant colonies were picked and sequentially subcultured on solid medium containing increasing concentrations of basta. Seventeen colonies resistant to basta at high concentration (150 μg mL−1) were grown in 100 mL TPM for 10 days. Metabolites were extracted and analysed by LCMS.
Six transformants produced DMB, ten showed a significant reduction in production and one did not produce any DMB or related compounds. The lack of compound production demonstrates that dmbS has a functional role in the synthesis of DMB. PCR analysis on the seventeen transformants using primers which amplify a fragment of dmbS resulted in the amplification of a 1,126 bp product demonstrating that the dmbSgene was still intact and ectopic integration had occurred. Therefore both the reduction in DMB concentration in some clones and the total lack of production in one can be attributed to PTGS.
Expression of dmbS in A. oryzae
To investigate the early stages in the biosynthesis of DMB, dmbS was expressed in the heterologous hostA. oryzae with and without its cognate enoyl reductase gene (dmbC). The expression vector pTAex3GS was first converted to a yeast-E. coli shuttle vector, pTAYAGSarg; this allowed assembly of the dmbS coding region by homologous recombination in yeast, replacing the Gateway destination sequence within the amyB expression cassette. The resultant plasmid, pTAYAarg·dmbS, was used to transform A. oryzaeM-2-3 (argB−), and colonies were selected on a minimal medium lacking arginine. Twenty six transformants were grown in 100 mL Czapek-Dox Maltose Peptone (CMP) medium for eight days, then the metabolites were extracted and analysed by LCMS.Two compounds were isolated from one of the transformants grown at 1.5 L scale using mass directed HPLCpurification targeted at 340 and 400 m/z units ([M]H+). The smaller of the two compounds (2.6 mg, 1.7 mg L−1) had a HRMS for [M − H]− of 338.1454 indicating a formula of C20H20NO4 (calc 338.1398). Both compounds displayed resonances in the 500 MHz 1H NMR indicative of a tyrosine-derived tetramic acid, confirmed by typical UV maxima at 390 nm, although they differed in the polyketide resonances. The smaller compound possesses four contiguous alkeneprotons and the lack of an HMBC correlation between the side-chain methyl and carbonylC-6 indicated it could be the desmethyl-prototenellin B analogue 8 (Fig. 4). This was then confirmed by full 2D NMR analysis.
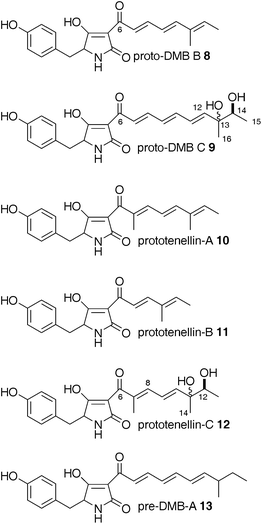 | ||
| Fig. 4 Tetramic acids observed in this study. | ||
The larger compound was unstable and isolated as an enriched mixture (3.2 mg, 2.1 mg L−1). 1H and COSYNMR indicated six contiguous olefinic protons, i.e. a –CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH– longer than its smaller counterpart 8, and a similarly located methyl group. ESI-MS indicated the presence of two hydroxyls. Full 13C NMR characterisation could not be made but HMBC analysis of positions 12 to 16, and comparison with prototenellin-C 1214 (vide infra) confirmed the structure as a 1
CH– longer than its smaller counterpart 8, and a similarly located methyl group. ESI-MS indicated the presence of two hydroxyls. Full 13C NMR characterisation could not be made but HMBC analysis of positions 12 to 16, and comparison with prototenellin-C 1214 (vide infra) confirmed the structure as a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 diastereomeric mixture of diols9. This presumably arises via stereoselective 13–14 epoxidation in the A. oryzaehost followed by acid catalysed hydrolysis during the extraction procedure (Scheme 2).
:1 diastereomeric mixture of diols9. This presumably arises via stereoselective 13–14 epoxidation in the A. oryzaehost followed by acid catalysed hydrolysis during the extraction procedure (Scheme 2).
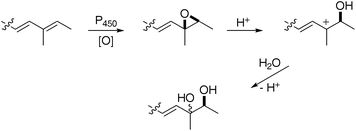 | ||
| Scheme 2 Formation of diols9 and 12 as mixtures of diastereomers. | ||
Effect of NRPS
We constructed a chimaeric PKS-NRPS consisting of the tenellin PKS fused to the DMB NRPS. The fusion site was chosen in a region between the ACP of TENS and the C-domain of DMBS. The chimaeric coding region was constructed by replacing the PKS portion of pTAYAarg·dmbS with its tenS counterpart by homologous recombination in yeast (Scheme 3).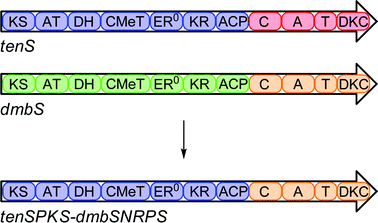 | ||
| Scheme 3 Construction of chimaeric PKS-NRPS. | ||
A. oryzaeM-2-3 was transformed with the resultant plasmid, pTAYAarg·tenSdmbS1, and 10 clones capable of growth on a minimal medium lacking arginine were selected. These were grown in CMP liquid medium for 7 days, and then metabolites were extracted and examined by LCMS. Seven clones showed the presence of compounds not observed in an untransformed A. oryzae control. Three compounds were observed which corresponded to compounds already known from the expression of tenS alone—prototenellin-A 10, prototenellin-B 11 and prototenellin-C 12. Surprisingly these clones appeared to be more productive than the original A. oryzae tenS expression clones described previously.6 In particular the compound identified as prototenellin-C 12, which had previously been only partially characterised due to lack of material (2.6 mg L−1 from pTAYAarg·tenS), was present in sufficient quantity from the chimeric clone pTAYAarg·tenSdmbS1 (9.6 mg L−1) to allow purification and structure elucidation.
Thus prototenellin-C 12 (m/z of 410, [M]Na+) was targeted for purification by mass-directed HPLC to afford 9.6 mg of pure material. HRMS (410.1578) indicated a molecular formula of C21H25NO6Na ([M]Na+, calc 410.1580). 1H NMR at 500 MHz revealed the presence of a para-substituted aromatic, a tetramic acid (coupled methine and diastereomeric methylene), and an unfamiliar polyketide side chain, consistent with the previous incomplete structural work. Signals from the polyketide side chain appeared to indicate the presence of two isomeric forms. Three contiguous olefinic protons indicated that the methyl groups must be separated, as in prototenellin-A 10. A singlet methyl at 1.92 ppm corresponded to C-15 and HMBC correlation to C-6 and C-8 also indicated that C-15 was attached to C-7. Observation of two methyl singlets at 1.28 and 1.30 ppm and two overlapping methyl doublets at 1.14 and 1.16 ppm, and the requirement to fit in two OHgroups, suggested the 1:1 mixture of diastereomers shown in Fig. 4. This structure was confirmed by full 13C and 2D NMR analysis and it presumably arises similarly to 9 (Scheme 2).
Effect of dmbC
The trans-actingERgenedmbC was then placed in the amyB expression cassette of pBarAmyBGS, in which a basta-resistance gene replaces the argB selectable marker of pTAex3GS. The resultant plasmid, pBardmbC, was used together with pTAYAdmbS to transform A. oryzaeM-2-3, and co-transformants were selected on minimal medium containing basta.15 Twenty four colonies were subcultured on solid medium containing an increased concentration of basta (150 μg mL−1). Five dmbC/dmbS cotransformants were grown and analysed by LCMS as before. One of these transformants produced a new compound which appeared to be an unsaturated tetramic acid because of its distinctive UV absorbance at 390 nm, similar to pretenellin-A 2, while its mass (368, [M]H+) indicated the presence of a single nitrogen atom.Mass-directed HPLCpurification was again used to purify the new compound (17.6 mg, 17.6 mg L−1). HRMS (368.1851, [M]H+) indicated a molecular formula of C22H26NO4 (calc 368.1862). 1H NMR analysis at 500 MHz indicated a near identical spectrum for the polyketide side chain as DMB5. Similarly, a para-substituted aromatic moiety was clearly present, but the distinctive pyridone H-6 singlet was missing and replaced by coupled methine and diastereotopic methyleneprotons characteristic of tetramic acids. Full 2D analysis and comparison with pretenellin-A 2 revealed the structure as the expected tetramic acid13 which we name preDMB-A.
In a parallel experiment A. oryzae was cotransformed with dmbS and tenC expression constructs. Ten of the arginine-independent, basta-resistant dmbS/tenC cotransformants were grown in liquid medium, and LCMS analysis showed that four of these produced preDMB-A 13 (23.8 mg L−1). This was confirmed by purification and 1H NMR analysis.
In a reciprocal experiment A. oryzaeM-2-3 was cotransformed with tenS and dmbC expression constructs. The six transformants obtained produced the previously characterised compound pretenellin-A 2 (25.0 mg L−1). Again, this was confirmed by purification and NMR analysis.
Discussion
The discovery of the origin of programming in fungal HR-PKS remains a key intellectual challenge. Solving the programming problem could potentially allow these biological synthases to be manipulated to produce new and useful compounds at will. There are currently three different paradigm HR-PKS systems known. The first, exemplified by squalestatin tetraketide synthase (SQTKS)16 and lovastatindiketide synthase (LDKS)17 features an iterative type I PKS in which the enoyl-reductase (ER) domain is clearly present and functional. The second type of iterative HR-PKS system, exemplified by lovastatin nonaketide synthase (LNKS),17tenellin synthetase (TENS)4 and aspyridone synthetase (APDS)18 has a defective enoyl reductase (ER0) domain which is complemented by a trans-actingER. In these three cases the PKS is also fused to a partial (LNKS), or full (TENS and APDS), NRPS module. Thirdly, systems such as the fusarin synthetase (FUSS)19 have a defective ER domain which is not complemented by any trans-actingprotein—such HR-PKS produce polyunsaturated chains.Vederas and coworkers were the first to report how the trans-acting ER can modulate the programme of the native HR-PKS.17 In the case of LNKS, co-expression of lovB (encoding LNKS) and lovC (encoding the trans-actingER) produces the nonaketide dihydromonacolin L 16a—a direct precursor of lovastatin17a. However expression of lovB alone leads to the production of abberant hexa- and hepta-ketide pyrones 14 and 15 (Scheme 4). Both pyrones are C-methylated after the third extension and so methylation is maintained despite the loss of enoylreduction and chain-length fidelity. In recent work Vederas, Tang and coworkers have reconstructed LNKS in vitro and replicated the in vivo results.20 Additionally they showed that replacement of the lovC ER by a different trans-acting ER from the compactin cluster led to no change in programming as dihydromonacolin-L16a was produced in the presence of SAM, although in the absence of SAM differences in programming were observed. In these experiments no product was formed when LNKS and the lovCER were incubated, but desmethylmonacolin-L16b was made when the compactin (17b) ER was used instead of the lovCER.
 | ||
| Scheme 4 Compounds observed as products of the lovastatin nonaketide synthase (LNKS) and the aspyridone (APDS) and cyclopiazonic acid (CPAS) synthetases. | ||
We have also observed comparable behaviour in the tenellin system. The TENS PKS-NRPS produces a range of aberrantly programmed tetramic acids. Three reprogrammed polyketides have been isolated and characterised after in vivo expression (prototenellins A to C 10 to 12, Fig. 4, Fig. 5 columns 1 and 2) while numerous likely methylation isomers have been detected by LCMS.6 However, when tenS is expressed with tenC a single compound is produced where the polyketide moiety is the same as in tenellin1 (Fig. 5, column 3).6
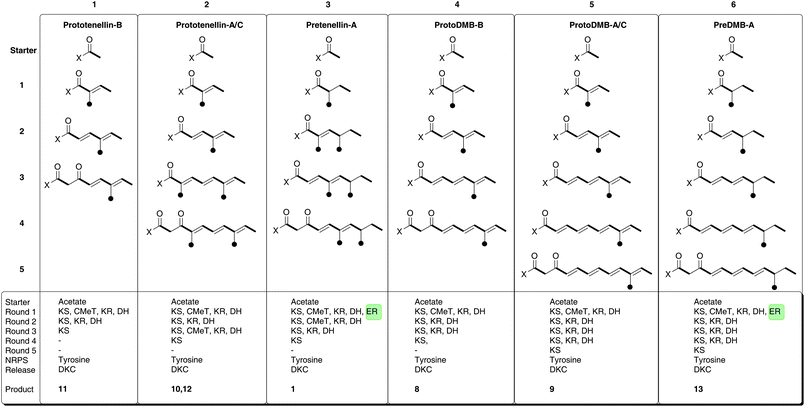 | ||
| Fig. 5 Programming evident during the operation of TENS and DMBS. | ||
We now report the activity of a HR-PKS related to TENS. The DMB synthetase (DMBS) is 86% identical at the peptide level to TENS (and up to 97% identical for individual domains)—but despite this it is programmed differently. The chain is one extension longer, but methylated only once. Like TENS, DMBS appears to show loose programming fidelity in the absence of its cognate trans-acting ER encoded by dmbC, synthesising the shorter chain compound 8 which was isolated and characterised (Fig. 5, column 4), and the expected full-length, fully unsaturated compound which is the precursor of diol9 (Fig. 5, column 5).
We constructed a chimaeric system consisting of the TENS PKS and the DMBS NRPS and the results suggest that the NRPS subunit plays no role in programming the PKS component. The TENS NRPS C-domain is evidently able to process the three different chains of 2, 10 and 11, while the DMBS NRPS C-domain can process all six different carbon chains shown in Fig. 5. Thus the C-domain appears to play no role in selecting a particular polyketide structure for condensation with tyrosine. This contrasts to the results of Tang and coworkers who have observed that the APDS NRPS module shows a strict substrate selectivity and appears not to accept incorrectly programmed polyketide intermediates, e.g. lacking C-methyl branches or containing unreduced alkenes in in vitro assays.21 Our observation supports a model in which the PKS is normally relatively fast, producing its programmed product which then awaits attachment to tyrosine by the relatively slow NRPS components. Thus the NRPS can be thought of as an effective off-loading mechanism for the PKS in the cases of TENS and DMBS. If the NRPS components were faster than the PKS then it should be possible for a catholic C-domain to select part-constructed polyketides for condensation with tyrosine which it does not appear to do in the presence of a trans-actingER. This model also suggests that when the trans-acting ER is absent the PKS becomes relatively slow, allowing the C-domain to select shorter polyketides as observed in the cases of 8 and 12. However, we have not observed the synthesis of longer chains by either TENS or DMBS.
Our observations may also explain some of the other reported results. When Tang and coworkers incubated the APDS PKS with an NRPS from the cyclopiazonic acid (21, Scheme 4) synthetase (CPAS) they observed the formation of the normal APDS tetraketide fused to tryptophan—clearly the CPAS NRPS C-domain does not intercept its usual diketide substrate but accepts the correctly elaborated polyketide synthesised by the APDS PKS. When APDS was incubated in vitro in the absence of SAM or the APDC trans-actingenoyl reductase, synthesis of longer (penta and hexa) polyketide pyrones were observed. In such cases it appears that the APDS PKS makes its initial product rapidly, but that it cannot be off-loaded because the NRPS is highly selective for its native polyketide intermediate (in contrast to TENS). The ACP-bound polyketide could then—slowly—be extended to form a diketo-thiolester which can self-release as a pyrone. This is exemplified when tyrosine is omitted from the assay which disables the NRPS, again resulting in the formation of a pyrone. Interestingly when the PKS portion of APDS is produced as a stand-alone protein and incubated with SAM, NADPH and the APDC trans-actingenoyl reductase, the normal tetraketide with the correct methylation and reduction programming is similarly further chain extended and released as a hexaketide pyrone.
We also addressed one of the key questions regarding programming in HR iterative PKS: what programming role do trans-actingERs play? Thus tenS and dmbS have been expressed with either tenC or dmbC. In all cases the compound produced was clearly directed by the PKS component rather than the ER component. This indicates that the PKS controls the programme in the presence of the trans-actingER. In the absence of the ER the PKS loses fidelity—with respect to chain length and methylation pattern—for the second methylation. This observation supports a model in which the trans-acting ER binds to the PKS-NRPS and perhaps modulates its activity through allosteric effects. Our results show that this both tightens the fidelity of the HR-PKS and also increases its efficiency as yields of compounds produced from the coexpression experiments were higher than those obtained when the PKS-NRPS was expressed alone. For example pre-DMBA 13 was produced at 23 mg L−1 and 17.6 mg L−1 when dmbS was coexpressed with dmbC or tenC respectively, vs. 3.8 mg L−1 total purified yield of proto-DMB B 8 and proto-DMB C 9 from the expression of dmbS alone. Likewise the yields of pretenellin A 2 were higher than of the combined yields of prototenellins A to C 10–12.6
The creation of the TENS-PKS:DMBS-NRPS hybrid system is the first report of an in vivo functional hybrid fungal PKS-NRPS. Bearing in mind that independent transformants express the same transgene to varying extents, the hybrid appears to be more active than either of its progenitors, producing purified yields of prototenellin C at 9.6 mg L−1vs. 2.4 mg L−1 (tenS) and 2.1 mg L−1 (dmbS). This is again in contrast to recent reports of in vitro fungal PKS-NRPS hybrids created from the APDS PKS and the CPAS NRPS which were very weakly active.21 The fact that the TENS-PKS:DMBS-NRPS hybrid is fully functional is an important observation as this suggests that the creation of in vivo hybrid HR-PKS systems may allow a more precise dissection of programming by these enzymes. Indeed, such work is currently under way in our laboratories.
Acknowledgements
This work was supported by BBSRC grant BB/E007791/1 to CML and EPSRC grant EP/F066104/1 to RJC. KW was supported by a BBSRC Doctoral Training Grant. The authors are also grateful for funding from: Kano State Government Nigeria, The MacArthur Foundation, Bayero University and the Nigerian Petroleum Technology Fund (AAY); Beni Suef University and the Egyptian Ministry of Higher Education (WB); Higher Education Commission of Pakistan (ZW); Majlis Amanah Rakyat (MARA) and Institute for Medical Research (IMR), Malaysia (KAK). We also thank James Lewis, Roger Read and Dr Laurel Russell (School of Biological Sciences) for technical assistance.Notes and references
- R. Geris and T. J. Simpson, Nat. Prod. Rep., 2009, 26, 1063–1094 RSC.
- R. J. Cox and T. J. Simpson, Complex Enzymes In Microbial Natural Product Biosynthesis, Part B: Polyketides, Aminocoumarins and Carbohydrates, Methods Enzymol., 2009, 459, 49–78 CAS.
- R. J. Cox, Org. Biomol. Chem., 2007, 5, 2010–2026 RSC.
- K. L. Eley, L. M. Halo, Z. Song, H. Powles, R. J. Cox, A. M. Bailey, C. M. Lazarus and T. J. Simpson, ChemBioChem, 2007, 8, 289–297 CrossRef CAS.
- L. M. Halo, M. N. Heneghan, A. A. Yakasai, Z. Song, K. Williams, A. M. Bailey, R. J. Cox, C. M. Lazarus and T. J. Simpson, J. Am. Chem. Soc., 2008, 130, 17988–17996 CrossRef CAS.
- L. M. Halo, J. W. Marshall, A. A. Yakasai, Z. Song, C. P. Butts, M. P. Crump, M. Heneghan, A. M. Bailey, T. J. Simpson, C. M. Lazarus and R. J. Cox, ChemBioChem, 2008, 9, 585–594 CrossRef CAS.
- M. N. Heneghan, A. A. Yakasai, L. M. Halo, Z. Song, A. M. Bailey, T. J. Simpson, R. J. Cox and C. M. Lazarus, ChemBioChem, 2010, 11, 1508–1512 CrossRef CAS.
- J. W. Sims and E. W. Schmidt, J. Am. Chem. Soc., 2008, 130, 11149–11155 CrossRef CAS; X. Liu and C. T. Walsh, Biochemistry, 2009, 48, 8746–8757 CrossRef CAS.
- A. G. McInnes, D. G. Smith, C. K. Wat, L. C. Vining and J. L. C. Wright, J. Chem. Soc., Chem. Commun., 1974, 281–282 RSC; C. K. Wat, A. G. McInnes, D. G. Smith, J. L. C. Wright and L. C. Vining, Can. J. Chem., 1977, 55, 4090–4098 CAS.
- S. Takahashi, N. Kakinuma, K. Uchida, R. Hashimoto, T. Yanagisawa and A. Nakagawa, J. Antibiot., 1998, 51, 596–598 CAS.
- J. H. Yu, Z. Hamari, K. H. Han, J. A. Seo, Y. Reyes-Dominguez and C. Scazzocchio, Fungal Genet. Biol., 2004, 41, 973–981 CrossRef CAS.
- M. N. Heneghan, A. M. Costa, M. P. Challen, P. R. Mills, A. Bailey and G. D. Foster, Mol. Biotechnol., 2007, 35, 283–296 Search PubMed.
- X. F. Zheng, Y. Kobayashi and M. Takeuchi, Appl. Microbiol. Biotechnol., 1998, 49, 39–44 CrossRef CAS.
- Naming Conventions: We refer to genuine precursors with the name prefix pre- these are compounds which lie on the biosynthetic route to the final compound. For example pretenellin-A 2 is a direct precursor of tenellin1. Compound names with the prefix proto- refer to shunt or reprogrammed metabolites which cannot be precursors—for example prototenellin-A 10 cannot be a precursor of tenellin1. In these cases A, B, C etc. refer to the order in which the compounds were discovered.
- Note that sometimes individual transformants do not sporulate well or are difficult to use for the preparation of protoplasts for the second transformation, so it can be better to do co-transformation to construct the dual expression systems. Other times the reverse can be true and stepwise transformation is more effective.
- R. J. Cox, F. Glod, D. Hurley, C. M. Lazarus, T. P. Nicholson, B. A. M. Rudd, T. J. Simpson, B. Wilkinson and Y. Zhang, Chem. Commun., 2004, 2260–2261 RSC.
- J. Kennedy, K. Auclair, S. G. Kendrew, C. Park, J. C. Vederas and C. R. Hutchinson, Science, 1999, 284, 1368–1372 CrossRef CAS.
- S. Bergmann, J. Schumann, K. Scherlach, C. Lange, A. A. Brakhage and C. Hertweck, Nat. Chem. Biol., 2007, 3, 213–217 CrossRef CAS.
- Z. Song, R. J. Cox, C. M. Lazarus and T. J. Simpson, ChemBioChem, 2004, 5, 1196–1203 CrossRef CAS.
- S. M. Ma, J. W. H. Li, J. W. Choi, H. Zhou, K. K. M. Lee, V. A. Moorthie, X. K. Xie, J. T. Kealey, N. A. Da Silva, J. C. Vederas and Y. Tang, Science, 2009, 326, 589–592 CrossRef CAS.
- W. Xu, X. Cai, M. E. Jung and Y. Tang, J. Am. Chem. Soc., 2010, 132, 13604–13607 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c1sc00023c |
| This journal is © The Royal Society of Chemistry 2011 |