Chemical tuning of CO2 sorption in robust nanoporous organic polymers†
Robert
Dawson
,
Dave J.
Adams
and
Andrew I.
Cooper
*
Department of Chemistry and Centre for Materials Discovery, University of Liverpool, Liverpool, United Kingdom. E-mail: aicooper@liv.ac.uk
First published on 1st April 2011
Abstract
We report here the carbon dioxide sorption properties for a series of conjugated microporous polymer (CMP) networks. These CMP materials incorporate a range of chemical functionalities including carboxylic acids, amines, hydroxyl groups, and methyl groups. The carboxylic acid functionalised network, rather than its amine analogue, shows the highest isosteric heat of sorption for CO2. This supports recent computational predictions for metal–organic frameworks and suggests that acid-functionalised frameworks could outperform more widely studied amine sorbents in CO2 capture and separation application.
Introduction
The capture of carbon dioxide produced via the burning of fossil fuels is an important challenge for society.1,2Carbon dioxide capture in porous adsorbents is one possible approach. The ideal properties for a CO2 adsorbent will depend on the intended point of use. In pre-combustion capture of CO2 from syngas (H2, CO2 and CO), or from natural gas reserves, the gas stream contains approximately 35.5% CO2 and requires materials to be used at relatively high temperatures and pressures. For post-combustion capture, the CO2 percentage in the gas stream is lower (15–16%) and there is also a higher percentage of water vapour. As such, long-term hydrolytic stability is a consideration. Many different materials have been investigated for carbon capture and storage (CCS). Amine-containing solvents have been explored for some years,3 but recycling these materials on a large scale for use in power plants may require 20–40% of the energy produced. This, in turn, has been estimated by some authors to increase the cost of energy production by 70%.2 Other materials investigated as sorbents include supported amines,4–7carbon-based sorbents,8–10zeolites11–13 and supported carbonates.14Microporous organic polymer networks15 combine high surface areas16 with good physicochemical stability and potential for synthetic diversification. Microporous polymer networks have been the investigated for applications such as catalysis,17,18 gas separation18,19 and gas storage,18,20–26 For example, the storage of hydrogen gas has been reported for a number of microporous materials21,22,27–31 although the volume of gas stored at room temperature is still far below the US Department of Energy targets.32 By contrast, CO2 gas interacts more strongly with most sorbents and displays isosteric heats of sorption in the broad range required for sorption close to ambient temperature.
CO2 sorption properties have been reported for a range of high surface area materials33 under varying conditions of temperature and pressure. For example, CO2 uptake in metal–organic frameworks (MOFs) is often reported at high pressures (10–50 bar)34–37 —that is, under conditions which are more relevant to pre-combusion capture.33 The post-combusion capture of CO2 might be problematic for some MOFs due their relative hydrolytic instability38 although zeolitic imidazolate frameworks (ZIFs),39–41 for example, are moisture stable and show good CO2 uptake at atmospheric pressure. As an example, ZIF-78 has an uptake of 2.3 mmol g−1CO2 at 298 K and 1 bar.39,42 Microporous organic polymers can also exhibit good physicochemical stability. It was shown recently that highly porous polyphenylene networks (SABET > 5000 m2 g−1) can be boiled in water for a week with no loss of porosity.16 There are, however, relatively a few reports of CO2 capture in microporous polymers16,22,43 and those studies, too, have mainly focused on high pressure sorption conditions.
For post-combustion CO2 capture at ambient pressure, materials with very high surface areas may not be optimal.44 Rather, increasing the heat of adsorption through the introduction of tailored binding functionalities could have more potential to increase the amount of gas adsorbed. Moreover, the ability to tune isosteric heats of sorption through chemical synthesis offers the potential to optimize the adsorption and desorption profiles of the sorbent in order to reduce energy consumption in pressure-swing or temperature swing processes.45 Recently, Torrisi et al. calculated the isosteric heats of sorption for CO2 in functionalised MOFs and suggested that the sorption enthalpy might be modified by choice of the functional group in the organic linker.46 They predicted that the incorporation of a carboxylic acid group would lead to the highest isosteric heat, challenging the current research emphasis in the literature regarding amine groups for CO2 capture.3,47–49 The synthetic incorporation of free acid functionalities, however, might be problematic in some MOF systems due to the propensity for these groups to coordinate with metals. On the other hand, we have previously reported the synthesis of a number of functionalised conjugated microporous polymer (CMP) networks50–54 synthesised viapalladium catalysed Sonogashira–Hagihara cross-coupling reactions. These networks can be synthesised with a wide range of pendant functionalities52,55 and with moderate to high Brunauer–Emmett–Teller (BET) surface areas. We report here the synthesis of two new networks which incorporate primary amine and carboxylic acid groups, respectively. We compare the isosteric heats and CO2 uptakes for these polymers in addition to other functionalised CMPs and discover a surprisingly close correlation between our materials and the aforementioned computational predictions for MOFs,46 suggesting perhaps a design principle which spans more than one class of material.
Results and discussion
Two novel functionalised conjugated polymer networks were synthesised using the Sonogashira–Hagihara palladium catalyzed cross-coupling reaction of 1,3,5-triethynylbenzene with either 2,5-dibromobenzoic acid or 2,5-dibromoaniline to yield the corresponding carboxylic acid- and amine-functionalized CMP networks, CMP-1-COOH and CMP-1-NH2, respectively (Scheme 1).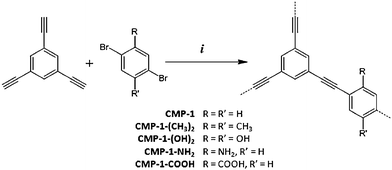 | ||
| Scheme 1 Synthesis of functionalised CMPs using (i) DMF, NEt3, Pd(PPh3)4, CuI, 100 °C, 72 h. | ||
The polymers were isolated as brown powders and analysed by FT-IR spectroscopy to show the presence of the acid and amine functionalities (Fig. S8†). A broad peak at 3421 cm−1 for CMP-1-COOH shows the presence of O–H in the acid together with a strong carbonyl stretch at 1707 cm−1. The N–H bonds in CMP-1-NH2 give rise to bands at 3466 and 3376 cm−1. Both networks show the presence of C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C functionalities at 2196 cm−1 as well as a smaller peak at 2107 and at 3294 cm−1 showing terminal alkyne groups (Fig. S8†). Further analysis of the networks by TGA showed that the networks were thermally stable to around 300 °C (see Fig. S1, Supporting Information†). The networks are also insensitive to treatment with water. Scanning electron microscopy showed that the powder morphology was similar to that reported for other CMP networks.55,56
C functionalities at 2196 cm−1 as well as a smaller peak at 2107 and at 3294 cm−1 showing terminal alkyne groups (Fig. S8†). Further analysis of the networks by TGA showed that the networks were thermally stable to around 300 °C (see Fig. S1, Supporting Information†). The networks are also insensitive to treatment with water. Scanning electron microscopy showed that the powder morphology was similar to that reported for other CMP networks.55,56
The CMP networks were analysed by nitrogen gas sorption. Adsorption/desorption isotherms (each offset for clarity by 100 cm3 g−1, Fig. 1. For non-offset data, see Fig. S2†) were collected at 77 K and showed a mainly Type I shape with high gas uptake at low pressures for networks CMP-1, CMP-1-NH2 and CMP-1-COOH with slight a slight hysteresis and Type IV isotherms with H2 hysteresis for networks CMP-1-(CH3)2 and CMP-1-(OH)2 as classified by IUPAC.57 The Brunauer–Emmett–Teller surface areas (SABET) were calculated from the adsorption branch of the nitrogen isotherms over a relative pressure range of P/P0 = 0.01–0.1 and were found to be 522 and 710 m2 g−1 for CMP-1-COOH and CMP-1-NH2 respectively. Micropore volumes were calculated using the t-plot method to be 0.16 and 0.24 cm3 g−1, while the total pore volumes of the two networks were 0.30 and 0.39 cm3 g−1. The surface areas and pore volumes of the five networks are compared in Table 1. CMP-1-COOH and CMP-1-NH2 have the two lowest BET surface areas in this series. To estimate the contribution of microporosity to the total porosity in the networks, we have calculated the ratio of V0.1/VTot, the ratio of the pore volume at P/P0 = 0.10 to the total pore volume at P/P0 = 0.99. CO2 gas sorption measurements were also obtained for the five networks at both 273 K and 298 K. The non-functionalised CMP-1 network showed the highest volumetric CO2 uptake at 298 K, adsorbing 1.18 mmol g−1 of CO2 at 1 bar. The dihydroxy network adsorbed less CO2 (1.07 mmol g−1 at 1 bar), despite exhibiting a higher surface area and pore volume. The dimethyl network, CMP-1-(CH3)2, showed the lowest uptake of CO2 (0.94 mmol g−1), despite having a higher surface area than CMP-1. As a comparison, BPL carbon (SABET = 1150 m2 g−1; a common reference material for CO2 uptake) exhibits an uptake of 1.9 mmol g−1. at 1 bar and 298 K.39,42 As an additional reference, the covalent organic framework, COF-102 (SABET = 3620 m2 g−1) shows the highest CO2 uptake among COFs at a pressure of 55 bar and 298 K,22 however the CO2 uptake at 1 bar and 273 K is 1.56 mmol g−1 —that is, less than the lowest uptake observed in this CMP series (CMP-1-COOH = 1.60 mmol g−1; see Fig S3, Supporting Information† for data collected at 273 K). We can therefore conclude that the CO2 uptake in these networks at lower pressures is not dependant soley on the surface area or pore volume. It would seem that the different chemical functional groups, as well as pore size, have a large effect on the uptake of gas in this pressure-temperature regime.
 | ||
| Fig. 1 Nitrogen adsorption (closed)/desorption (open) isotherms (77 K) for CMP-1 (black), CMP-1-(CH3)2 (green), CMP-1-(OH)2 (orange), CMP-1-NH2 (blue) and CMP-1-COOH (red) each offset by 100 cm3 g−1 for clarity. | ||
| Network | Ra | R′a | SA BET (m2 g−1)b | V micro (cm3 g−1)c | V total (cm3 g−1)d | V 0.1/tot |
|---|---|---|---|---|---|---|
| a R and R′ from Scheme 1. b Calculated over the relative pressure range P/P0 = 0.01–0.1. c Pore volume at P/P0 = 0.1. d Total pore volume (P/P0 = 0.99). e From ref. 56 | ||||||
| CMP-1 e | H | H | 837 | 0.32 | 0.45 | 0.71 |
| CMP-1-COOH | COOH | H | 522 | 0.22 | 0.30 | 0.73 |
| CMP-1-NH 2 | NH2 | H | 710 | 0.27 | 0.39 | 0.69 |
| CMP-1-(CH 3 ) 2 e | CH3 | CH3 | 899 | 0.34 | 0.75 | 0.45 |
| CMP-1-(OH) 2 e | OH | OH | 1043 | 0.40 | 0.71 | 0.56 |
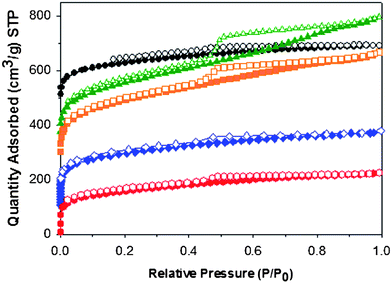
The isosteric heats of adsorption were calculated from the CO2 isotherms measured at 273 K and 298 K.20,26 The experimental isosteric heats showed the following order in terms of appended functional groups (Fig. 2d): –COOH > (OH)2 > NH2 > H > (CH3)2 at least for adsorbed gas quantities greater than 0.2 mmol g−1. At low absorption values, CMP-1-COOH shows a heat of adsorption reaching 33 kJ mol−1, dropping to around 27 kJ mol−1 at a pressure of 1 bar — that is, substantially higher than the less polar CMP materials in this series. The variation in CO2 sorption could potentially also arise from a difference in pore size distribution, with small pore sizes known to increase the heat of adsorption.31 Some differences in the pore size distribution were measured with N2 as the probe gas (Fig. S4†). However, using CO2 (which allows measurement of the smaller pores)58,59 the pore size distributions for all of these materials were found to be bimodal and very similar (Fig. S5†).
 | ||
| Fig. 2 (a) Calculated CO2 isotherms at 298 K for substituted MIL-53 frameworks, redrawn from ref. 46 and (b) measured CO2 isotherms for CMP networks. (c) Calculated isosteric heats of adsorption for CO2 in substituted MIL-53 frameworks, redrawn from ref. 46 and (d) measured for CMP networks. Colour-coding is as follows: unsubstituted networks (black); –(CH3)2 (green); –(OH)2 (orange); –NH2 (blue) and –COOH (red). | ||
As noted above, recent computational studies for a range of functionalised MOFs based on MIL-53 predicted that functional group modification would increase the amount of CO2 captured by the network.46 The calculations suggested that polar groups were effective in increasing CO2 capture, while bulky non-polar groups had a negative impact. It was predicted that the isosteric heat of adsorption would be highest for the carboxylic acid functionalised MIL, with the isosteric heats for the series to be in the order –COOH > –(OH)2 > –NH2 > –(CH3)2 > non-functionalized. For the carboxylic acid case, the CO2 was stabilised in the adsorption site by the presence of interactions from neighbouring carbonyl groups. Despite the fact that our CMP networks are quite different materials (amorphous not crystalline; covalent not metal–organic), we observed essentially the same trends in isosteric heats as predicted for the MIL-53 analogues46 (cf., Fig. 1c & 1d), thus supporting these calculations and perhaps suggesting a design principle which holds for more than one class of material. While it is not possible to fully deconvolute the effects of pore size (Fig. S4†) from functional group effects, our data corroborate the fundamental computational conclusion46 that carboxylic acid functionalities are a good target for CO2 capture materials. At this stage, we cannot say whether the adsorbed CO2 binds cooperatively to more than one –COOH group in the CMP network, as postulated for the MIL-53 system,46 since we do not have an unambigous structure model for these amorphous networks. It is likely however that there is a broader distribution of sorption sites in these amorphous networks, with varying propensities for multiple guest binding, and this may in part account for the broader range of isosteric heats as a function of coverage (Fig. 2d) for the experimental measurements with respect to the calculations which refer to perfectly ordered crystalline solids (Fig. 2c).46
By comparison with other materials, the heats of adsorption measured experimentally for CMP-1-COOH (32.6–26.1 kJ mol−1) are 15–20% higher than those reported by Luet al.43 for the network PPN-1. These values are also higher than activated carbon, but lower than some MOF networks. For example, one of the highest reported heats of adsorption for CO2 in MOFs (90 kJ mol−1) occurs in the material HCu[(Cu4Cl)3(BTTri)8(en)5] where a exposed metal centre binds to an ethylene diamine ligand which in turn binds to the CO2.60 This binding may in fact be too strong for capture applications and might translate to a large energetic penalty. Another material with a high heat of adsorption for CO2 is MIL-100, which contains open metal centres and exhibits a sorption enthalpy of 62 kJ mol−1 at low coverage.36 By contrast, the carbon material, BPL carbon, exhibits an isosteric heat of 24.3 kJ mol−1.61
Conclusions
We show here that chemical composition, much more than surface area, dictates the CO2 uptake for polymer networks at low pressures (1 atm) and close-to-ambient temperatures. The ability to fine-tune CO2 affinity in this way is of potential value for both CCS as well as applications such as gas separation. Our results validate previous computational predictions for MOF materials,46 suggesting that such ab initio computational studies have translational value within this class of materials. Our results, along with these previous calculations, also suggest that we should consider polar acidic functionalities in microporous frameworks and that such functionalities, as demonstrated here, might outperform aromatic amine functionalities for CO2 sorption.Acknowledgements
The authors would like to thank the Engineering and Physical Sciences Research Council (EP/G061785/1) for funding. A. I. C. is a Royal Society Wolfson Merit Award holder. The authors gratefully acknowledge Drs A. Torrisi, R. G. Bell and C. Mellot-Draznieks at UCL for allowing the reproduction of their data (ref. 46).Notes and references
- S. Chu, Science, 2009, 325, 1599 CrossRef CAS.
- R. S. Haszeldine, Science, 2009, 325, 1647–1652 CrossRef CAS.
- G. T. Rochelle, Science, 2009, 325, 1652–1654 CrossRef CAS.
- X. Xu, C. Song, J. M. Andrésen, B. G. Miller and A. W. Scaroni, Microporous Mesoporous Mater., 2003, 62, 29–45 CrossRef CAS.
- G. P. Knowles, J. V. Graham, S. W. Delaney and A. L. Chaffee, Fuel Process. Technol., 2005, 86, 1435–1448 CrossRef CAS.
- M. L. Gray, Y. Soong, K. J. Champagne, J. Baltrus, R. W. Stevens, P. Toochinda and S. S. C. Chuang, Sep. Purif. Technol., 2004, 35, 31–36 CrossRef CAS.
- T. Filburn, J. J. Helble and R. A. Weiss, Ind. Eng. Chem. Res., 2005, 44, 1542–1546 CrossRef CAS.
- M. G. Plaza, C. Pevida, A. Arenillas, F. Rubiera and J. J. Pis, Fuel, 2007, 86, 2204–2212 CrossRef CAS.
- C. Pevida, M. G. Plaza, B. Arias, J. Fermoso, F. Rubiera and J. J. Pis, Appl. Surf. Sci., 2008, 254, 7165–7172 CrossRef CAS.
- C. Pevida, T. C. Drage and C. E. Snape, Carbon, 2008, 46, 1464–1474 CrossRef CAS.
- J. Merel, M. Clausse and F. Meunier, Ind. Eng. Chem. Res., 2008, 47, 209–215 CrossRef CAS.
- D. Ko, R. Siriwardane and L. T. Biegler, Ind. Eng. Chem. Res., 2003, 42, 339–348 CrossRef CAS.
- S. Cavenati, C. A. Grande and A. E. Rodrigues, J. Chem. Eng. Data, 2004, 49, 1095–1101 CrossRef CAS.
- N. Shigemoto, T. Yanagihara, S. Sugiyama and H. Hayashi, Energy Fuels, 2006, 20, 721–726 CrossRef CAS.
- A. I. Cooper, Adv. Mater., 2009, 21, 1291–1295 CrossRef CAS.
- T. Ben, H. Ren, S. Q. Ma, D. P. Cao, J. H. Lan, X. F. Jing, W. C. Wang, J. Xu, F. Deng, J. M. Simmons, S. L. Qiu and G. S. Zhu, Angew. Chem., Int. Ed., 2009, 48, 9457–9460 CrossRef CAS.
- J. Schmidt, J. Weber, J. D. Epping, M. Antonietti and A. Thomas, Adv. Mater., 2009, 21, 702–705 CrossRef CAS.
- N. B. McKeown and P. M. Budd, Chem. Soc. Rev., 2006, 35, 675–683 RSC.
- P. M. Budd and N. B. McKeown, Polym. Chem., 2010, 1, 63–68 RSC.
- C. D. Wood, B. Tan, A. Trewin, H. J. Niu, D. Bradshaw, M. J. Rosseinsky, Y. Z. Khimyak, N. L. Campbell, R. Kirk, E. Stockel and A. I. Cooper, Chem. Mater., 2007, 19, 2034–2048 CrossRef CAS.
- P. M. Budd, A. Butler, J. Selbie, K. Mahmood, N. B. McKeown, B. Ghanem, K. Msayib, D. Book and A. Walton, Phys. Chem. Chem. Phys., 2007, 9, 1802–1808 RSC.
- H. Furukawa and O. M. Yaghi, J. Am. Chem. Soc., 2009, 131, 8875–8883 CrossRef CAS.
- J. Germain, J. M. J. Fréchet and F. Svec, J. Mater. Chem., 2007, 17, 4989–4997 RSC.
- J. Germain, J. M. J. Fréchet and F. Svec, Chem. Commun., 2009, 1526–1528 RSC.
- B. S. Ghanem, K. J. Msayib, N. B. McKeown, K. D. M. Harris, Z. Pan, P. M. Budd, A. Butler, J. Selbie, D. Book and A. Walton, Chem. Commun., 2007, 67–69 RSC.
- C. D. Wood, B. Tan, A. Trewin, F. Su, M. J. Rosseinsky, D. Bradshaw, Y. Sun, L. Zhou and A. I. Cooper, Adv. Mater., 2008, 20, 1916–1921 CrossRef CAS.
- D. J. Collins and H. C. Zhou, J. Mater. Chem., 2007, 17, 3154–3160 RSC.
- N. B. McKeown, B. Gahnem, K. J. Msayib, P. M. Budd, C. E. Tattershall, K. Mahmood, S. Tan, D. Book, H. W. Langmi and A. Walton, Angew. Chem., Int. Ed., 2006, 45, 1804–1807 CrossRef.
- R. E. Morris and P. S. Wheatley, Angew. Chem., Int. Ed., 2008, 47, 4966–4981 CrossRef.
- L. J. Murray, M. Dinca and J. R. Long, Chem. Soc. Rev., 2009, 38, 1294–1314 RSC.
- F. Svec, J. Germain and J. M. J. Fréchet, Small, 2009, 5, 1098–1111 CrossRef CAS.
- S. Satyapal, J. Petrovic, C. Read, G. Thomas and G. Ordaz, Catal. Today, 2007, 120, 246–256 CrossRef CAS.
- D. D'Alessandro, B. Smit and J. Long, Angew. Chem., Int. Ed., 2010, 49, 6058–6082 CrossRef CAS.
- A. R. Millward and O. M. Yaghi, J. Am. Chem. Soc., 2005, 127, 17998–17999 CrossRef CAS.
- S. Couck, J. F. M. Denayer, G. V. Baron, T. Rémy, J. Gascon and F. Kapteijn, J. Am. Chem. Soc., 2009, 131, 6326–6327 CrossRef CAS.
- P. L. Llewellyn, S. Bourrelly, C. Serre, A. Vimont, M. Daturi, L. Hamon, G. De Weireld, J.-S. Chang, D.-Y. Hong, Y. Kyu Hwang, S. Hwa Jhung and G. R. Férey, Langmuir, 2008, 24, 7245–7250 CrossRef.
- H.-S. Choi and M. P. Suh, Angew. Chem., Int. Ed., 2009, 48, 6865–6869 CrossRef CAS.
- S. S. Kaye, A. Dailly, O. M. Yaghi and J. R. Long, J. Am. Chem. Soc., 2007, 129, 14176–14177 CrossRef CAS.
- A. Phan, C. J. Doonan, F. J. Uribe-Romo, C. B. Knobler, M. O'Keeffe and O. M. Yaghi, Acc. Chem. Res., 2010, 43, 58–67 CrossRef CAS.
- R. Banerjee, A. Phan, B. Wang, C. Knobler, H. Furukawa, M. O'Keeffe and O. M. Yaghi, Science, 2008, 319, 939–943 CrossRef CAS.
- H. Hayashi, A. P. Cote, H. Furukawa, M. O'Keeffe and O. M. Yaghi, Nat. Mater., 2007, 6, 501–506 CrossRef CAS.
- R. Banerjee, H. Furukawa, D. Britt, C. Knobler, M. O'Keeffe and O. M. Yaghi, J. Am. Chem. Soc., 2009, 131, 3875–3877 CrossRef CAS.
- W. Lu, D. Yuan, D. Zhao, C. I. Schilling, O. Plietzsch, T. Muller, S. Bräse, J. Guenther, J. Blümel, R. Krishna, Z. Li and H.-C. Zhou, Chem. Mater., 2010, 22, 5964–5972 CrossRef CAS.
- J. R. Holst and A. I. Cooper, Adv. Mater., 2010, 22, 5212–5216 CrossRef CAS.
- D. Aaron and C. Tsouris, Sep. Sci. Technol., 2005, 40, 321–348 CrossRef CAS.
- A. Torrisi, R. G. Bell and C. Mellot-Draznieks, Cryst. Growth Des., 2010, 10, 2839–2841 CrossRef CAS.
- N. Hiyoshi, K. Yogo and T. Yashima, Microporous Mesoporous Mater., 2005, 84, 357–365 CrossRef CAS.
- V. Zelenak, D. Halamova, L. Gaberova, E. Bloch and P. Llewellyn, Microporous Mesoporous Mater., 2008, 116, 358–364 CrossRef CAS.
- J. C. Hicks, J. H. Drese, D. J. Fauth, M. L. Gray, G. Qi and C. W. Jones, J. Am. Chem. Soc., 2008, 130, 2902–2903 CrossRef CAS.
- J. X. Jiang, F. Su, A. Trewin, C. D. Wood, N. L. Campbell, H. Niu, C. Dickinson, A. Y. Ganin, M. J. Rosseinsky, Y. Z. Khimyak and A. I. Cooper, Angew. Chem., Int. Ed., 2007, 46, 8574–8578 CrossRef.
- J.-X. Jiang, F. Su, A. Trewin, C. D. Wood, H. Niu, J. T. A. Jones, Y. Z. Khimyak and A. I. Cooper, J. Am. Chem. Soc., 2008, 130, 7710–7720 CrossRef CAS.
- J.-X. Jiang, A. Trewin, F. Su, C. D. Wood, H. Niu, J. T. A. Jones, Y. Z. Khimyak and A. I. Cooper, Macromolecules, 2009, 42, 2658–2666 CrossRef CAS.
- E. Stöckel, X. F. Wu, A. Trewin, C. D. Wood, R. Clowes, N. L. Campbell, J. T. A. Jones, Y. Z. Khimyak, D. J. Adams and A. I. Cooper, Chem. Commun., 2009, 212–214 RSC.
- J.-X. Jiang, A. Laybourn, R. Clowes, Y. Z. Khimyak, J. Bacsa, S. J. Higgins, D. J. Adams and A. I. Cooper, Macromolecules, 2010, 43, 7577–7582 CrossRef CAS.
- R. Dawson, A. Laybourn, R. Clowes, Y. Z. Khimyak, D. J. Adams and A. I. Cooper, Macromolecules, 2009, 42, 8809–8816 CrossRef CAS.
- R. Dawson, A. Laybourn, Y. Z. Khimyak, D. J. Adams and A. I. Cooper, Macromolecules, 2010, 43, 8524–8530 CrossRef CAS.
- K. S. W. Sing, D. H. Everett, R. A. W. Haul, L. Moscou, R. A. Pierotti, J. Rouquerol and T. Siemieniewska, Pure Appl. Chem., 1985, 57, 603–619 CrossRef CAS.
- D. Lozano-Castelló, D. Cazorla-Amorós and A. Linares-Solano, Carbon, 2004, 42, 1233–1242 CrossRef CAS.
- J. Weber, J. Schmidt, A. Thomas and W. Böhlmann, Langmuir, 2010, 26, 15650–15656 CrossRef CAS.
- A. Demessence, D. M. D'Alessandro, M. L. Foo and J. R. Long, J. Am. Chem. Soc., 2009, 131, 8784–8786 CrossRef CAS.
- K. B. Lee, M. G. Beaver, H. S. Caram and S. Sircar, Ind. Eng. Chem. Res., 2008, 47, 8048–8062 CrossRef CAS.
Footnote |
| † Electronic Supplementary Information (ESI) available containing the IR spectra, TGA, pore size distributions and CO2 adsorption isotherms See DOI: 10.1039/c1sc00100k/ |
| This journal is © The Royal Society of Chemistry 2011 |