Surface plasmon resonance imaging of glycoarrays identifies novel and unnatural carbohydrate-based ligands for potential ricin sensor development†
Margherita
Fais‡
ab,
Rositsa
Karamanska‡
ab,
Sarah
Allman
c,
Shirley A.
Fairhurst
a,
Paolo
Innocenti
c,
Antony J.
Fairbanks§
c,
Timothy J.
Donohoe
c,
Benjamin G.
Davis
c,
David A.
Russell
b and
Robert A.
Field
*ab
aDepartment of Biological Chemistry, John Innes Centre, Norwich Research Park, Norwich, NR4 7UH, UK. E-mail: rob.field@bbsrc.ac.uk
bSchool of Chemistry, University of East Anglia, Norwich Research Park, Norwich, NR4 7TJ, UK
cDepartment of Chemistry, University of Oxford, Chemistry Research Laboratory, Mansfield Road, Oxford, OX1 3TA, UK
First published on 28th July 2011
Abstract
Carbohydrate microarrays provide access to high through-put analysis of protein–carbohydrate interactions. Herein we demonstrate the use of SPR imaging (SPRi) of glycoarrays to assess the ligand specificity of the reputedly galactose-specific plant lectin RCA120 (Ricinus communis agglutinin 120), a surrogate for the bioterrorism agent ricin. Glycoarray studies identified RCA120 ligands based on galactose substituted at the 6-position with sialic acid. These observations, which were confirmed by saturation transfer difference (STD) NMR spectroscopy studies, inspired the synthesis of non-natural 6-substituted galactose derivatives, which were shown to have ∼3–4 fold enhanced binding to RCA120 with respect to the unsubstituted compound. These novel unnatural galactosides, which are chemically and biologically more robust than their natural glycan counterparts, represent new potential ligands for the development of carbohydrate-based ricin sensors.
Introduction
Major advances in analytical glycoscience over the past decade or so present many new opportunities for identifying carbohydrate–ligand interactions of biological significance.1–4 In particular, the development of carbohydrate and lectin microarrays5–9 provides access to quantitative, high throughput screening approaches that could not previously have been contemplated. In addition to the identification of natural ligands, these technologies also provide a basis for the rapid optimisation of non-natural ligands for carbohydrate-binding proteins, as exemplified by work on novel sialic acid derivatives,10 some of which serve as high affinity ligands for sialic acid-binding lectins (Siglecs).11 In the course of work on the development of ricin sensors,12 we had cause to consider the identification of novel ligands, and hence turned to carbohydrate microarrays.13Ricin is a lethal, ribosome-inactivating protein14,15 found in plants that are used for castor oil production. Its lectin-like B-chain facilitates adhesion of the toxin to non-reducing terminal galactose residues of glycoproteins on mammalian cell surfaces, resulting in the delivery of the toxic A-chain warhead into the cytosol. The A-chain catalyses the hydrolytic cleavage of a single base from eukaryotic ribosomal RNA, leading to a shutdown in protein synthesis and ultimately cell death. Ricin is several thousand times more toxic to man than cyanide, with the lethal dose for an adult estimated to be around 500 ng Kg−1 of body weight.14,15 Due to its high toxicity and accessibility, its potential as a biowarfare agent has long been recognised, with several recent incidents in the UK and the US resulting in threats to public safety.14,15 While recent efforts have identified potential leads for the development of anti-ricin inhibitors,16 a rapid method for the early detection of traces of the toxin remains an important goal.¶ Given the carbohydrate-binding properties of ricin B-chain, carbohydrate-based sensors are potentially attractive. Certain systems have been shown to exhibit very high selectivity for bacterial toxins17 and plant lectins, including ricin;12,18,19 related carbohydrate-based sensing platforms have also been used for bacterial detection.20–25 In addition, galactose-coated gold nanoparticles in a colorimetric assay12 for the detection ofRicinus communis agglutinin (RCA120), a surrogate for ricin (RCA60),26 afforded higher sensitivity than antibody-conjugated nanoparticles in piezoelectric immunosensing.27
Despite these early developments, D-galactosides are prevalent in nature and many other mammalian, plant and microbial lectins and enzymes recognise and/or metabolise this common monosaccharide.28 Selectivity is therefore a primary issue. In the context of robust ricin sensor development, we were drawn to explore carbohydrate microarrays in the search for alternative naturally occurring ligands for ricin that might provide insight for the design of non-natural ligands that in turn could show improved selectivity. We noted that surface plasmon resonance approaches had already been used successfully in conjunction with D-galactoside surfaces to develop biosensors for ricin.18,19 With a view to exploring a much wider range of potential toxin ligands, we were drawn to explore surface plasmon resonance imaging (SPRi), an array-based approach that is finding wide use in bimolecular interaction analysis and for the development of affinity-based biosensors.29 We have recently employed SPRi for the analysis of protein interactions with carbohydrate microarrays.13 Here we report the further use of this technique to analyse the interaction of RCA120 with an array of natural glycans, which in turn informed studies on ligand (re)design to provide unnatural sugar-based ligands for potential use in ricin sensors.
Results and discussion
Analysis of RCA120 interaction with a carbohydrate microarray by SPR imaging
Array generation through split-pin, contact printing of a library of 40 biotinylated glycans|| (Table 1) onto commercial neutravidin-coated gold sensor chips was carried out as reported previously.13 To briefly summarise, the library assessed was composed of a range of glycans with non-reducing termini based on: Gal-β-1,4 (7 compounds), Gal-β-1,3 (3), GalNAc (1), GlcNAc (4), sulfo-Gal (3), NeuAc-2,3 (12), NeuGc-2,3/2,6 (2/1), NeuAc-2,6 (4), and NeuAc-2,8 (3) (Table 1). The array was used to screen for RCA120 binders in the anticipation that glycans possessing a non-reducing terminal β-linked galactose residue (black and orange), the previously determined simple ligand for this lectin, would stand out as hits.13,18,19 Data for equilibrium binding of RCA120 and the α-2,6-NeuAc-specific Sambucus nigra agglutinin (SNA) to the glycoarray are presented in Fig. 1.**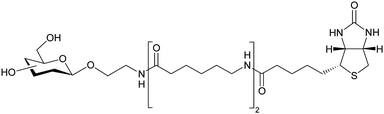
| Group name | Glycotope structure | Common name |
|---|---|---|
| B301 | Galβ1-4GlcNAcβ1-3Galβ1-3GlcNAc | L-NLec |
| B266 | Galβ1-4[Fucα1-3]GlcNAc | Lex |
| B186 | Galβ1-4(6OSO3)Glc | 6SuLac |
| B115 | Galβ1-4GlcNAcβ1-3Galβ1-4Glc | LNnT |
| B111 | (Galβ1-4GlcNAcβ)2 | Di-LN |
| B81 | Galβ1-4GlcNAc | LN |
| B80 | Galβ1-4Glc | Lac |
| B82 | Galβ1-3GlcNAc | Lec |
| B156 | Galβ1-3[Fucα1-4]GlcNAc | Lea |
| B277 | Galβ1-3GlcNAcβ1-3Galβ1-4GlcNAc | Lec-LN |
| B180 | GalNAcβ1-4GlcNAc | LDN |
| B299 | GlcNAcβ1-3Galβ1-3GlcNAc | GN-Lec |
| B181 | GlcNAcβ1-3Galβ1-4GlcNAc | GN-LN |
| B121 | GlcNAc | GN |
| B114 | GlcNAcβ1-3Galβ1-4Glc | LNT2 |
| B197 | [3OSO3]Galβ1-4Glc | 3′SuLac |
| B187 | [3OSO3]Galβ1-4(6OSO3)Glc | 6,3′-di-SuLac |
| B185 | (6OSO3)Galβ1-4Glc | 6′SuLac |
| B298 | Neu5Acα2-3[Neu5Acα2-3Galβ1-3GalNAcβ1-4]Galβ1-4Glc | GD4 |
| B273 | Neu5Acα2-3GalNAcβ1-4GlcNAc | 3′SLDN |
| B204 | Neu5Acα2-3[Galβ1-3GalNAcβ1-4]Galβ1-4GlcNAc | GM2(NAc) |
| B202 | Neu5Acα2-3[Galβ1-3GlcNAcβ1-4]Galβ1-4Glc | GM1 |
| B194 | Neu5Acα2-3[Galβ1-4GlcNAcβ1-3]3 | 3′SLN-LN-LN |
| B178 | Neu5Acα2-3[Galβ1-4GlcNAcβ]2 | 3′-SLN-LN |
| B177 | Neu5Acα2-3[GalNAcβ1-4]Galβ1-4Glc | GM2 |
| B174 | Neu5Acα2-3Galβ1-3[Fucα1-4]GlcNAc | SLea |
| B157 | Neu5Acα2-3Galβ1-4[Fucα1-3]GlcNAc | SLex |
| B85 | Neu5Acα2-3Galβ1-3GlcNAc | 3′SLec |
| B84 | Neu5Acα2-3Galβ1-4GlcNAc | 3′SLN |
| B83 | Neu5Acα2-3Galβ1-4Glc | 3′SLac |
| B93 | Neu5Gcα2-6Galβ1-4GlcNAc | 6′S(Gc)LN |
| B90 | Neu5Gcα2-3Galβ1-4GlcNAc | 3′S(Gc)LN |
| B89 | Neu5Gcα2-3Galβ1-4Glc | 3′S(Gc)Lac |
| B274 | Neu5Acα2-6GalNAcβ1-4GlcNAc | 6′SLDN |
| B179 | Neu5Acα2-6[Galβ1-4GlcNAcβ]2 | 6′-SLN-LN |
| B87 | Neu5Acα2-6Galβ1-4GlcNAc | 6′SLN |
| B86 | Neu5Acα2-6Galβ1-4Glc | 6′SLac |
| B184 | Neu5Acα2-8Neu5Acα2-3[GalNAcβ1-4]Galβ1-4Glc | GD2 |
| B108 | Neu5Acα2-8Neu5Acα2-8Neu5Acα2-3Galβ1-4Glc | GT3 |
| B107 | Neu5Acα2-8Neu5Acα2-3Galβ1-4Glc | GD3 |
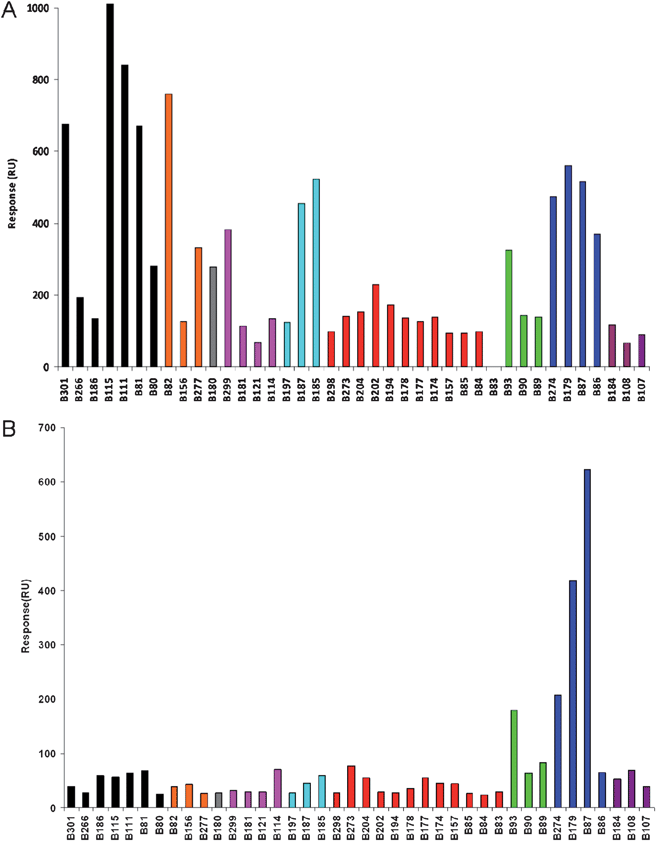 | ||
| Fig. 1 SPR imaging responses for equilibrium binding of (A) RCA120 (800 nM) and (B) SNA (80 nM) to an array of 40 biotinylated natural glycans (Table 1). Data errors were typically in the ±10–15% range.13 | ||
It was immediately evident from these array data that RCA120 had a preference for binding to non-reducing β-D-galactose (strongest responses are black and orange bars). However, such recognition was context-dependent, with Galβ1-4GlcNAc (B81) giving a stronger response than Galβ1-4Glc (B80), for instance; an extended glycan chain (e.g.B115) also gave a stronger response than the shorter counterpart (B81). Some galactose-terminated structures were relatively poorly recognised, in particular branched glycans, such as Lextrisaccharide (B266) and Lea trisaccharide (B156). As expected, RCA120 did not bind significantly to sugars with non-reducing terminal GlcNAc (pink) but results with sialic acid (NeuAc)-containing ligands were intriguing (red, green, blue, purple bars). On the whole, glycans terminated in α2-3Neu5Ac/Gc (red bars, plus B89, B90) were poor ligands whilst those terminated in α2-6Neu5Ac/Gc (blue bars, plus B93) showed relatively good binding to RCA120. Extending the α2-3Neu5Ac arm with an additional α2-8Neu5Ac residue did not recover binding (purple bars). These data are consistent with the ability of RCA120 to accommodate substitution at the 6-position of the non-reducing terminal β-galactose with sialic acid (Fig. 2); likewise, terminal galactose 6-sulfate was recognised by RCA120 whereas galactose 3-sulfate was not (cyan bars, B185 and B197, respectively). Together these data comprehensively map novel functional ‘hotspots’ for RCA120 carbohydrate binding.
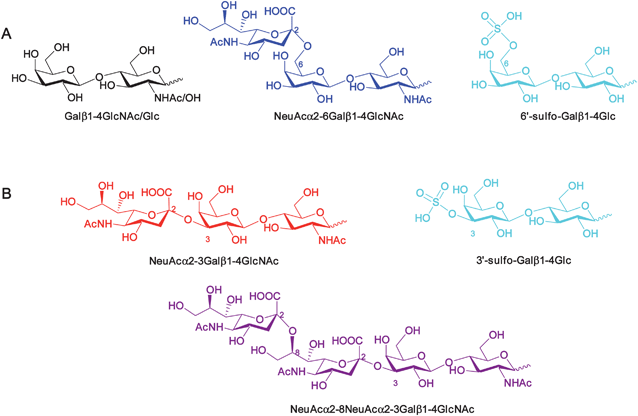 | ||
| Fig. 2 Structures of representative oligosaccharides identified from SPRi array analysis that (A) serve as ligands for RCA120 or (B) do not bind RCA120. Colour coding refers to Table 1/Fig. 1. | ||
Analysis of SNA interaction with a carbohydrate microarray by SPR imaging
Given these unexpected interactions of RCA120 with α-2,6-NeuAc-containing glycans, the glycoarray was probed with another previously characterised α-2,6-NeuAc-specific lectin, in this case Sambucus nigra agglutinin (SNA) (Fig. 1B). For SNA, the only glycan ligands that were identified in this study were three of the four α-2,6-NeuAc compounds (B87, B179, B274) and the single α-2,6-NeuGc (B93) out of the library of forty glycans. Together, these data confirmed the functional performance of the array bearing this key ligand type.Development of novel carbohydrate-based ligands for RCA120
The novel glycoarray data above revealed 6-modified D-galactosides as good RCA120 ligands. To explore the potential of these observations in the creation of bespoke ricin ligands a series of Galβ1,4-GlcNAc derivatives modified at the galactose 6-position were designed. The intention here was to attempt to move away from the potentially biologically and chemically labile α-2,6-linked NeuAc compounds in favour of a more robust ligand, which would be necessary for sensor applications. A library of Galβ1,4-GlcNAc derivatives modified at the galactose 6-position (1–7) was devised (Scheme 1), with a view to post-synthesis biotinylation through hydrazone chemistry32,33 (giving 1Bt–7Bt) to enable SPRi array analysis, as above. | ||
Scheme 1
Reagents and conditions: a) BF3·OEt2, DCM, −40 °C; b) 1. BF3·OEt2, CH3CN. 2. MeONa, MeOH. 3. H2, Pd(OH)2/C, MeOH for target compounds 1–4; b) 1. PPh3, H2O; 2. Ac2O, Py; 3. BF3·OEt2; 4. MeONa, MeOH; 5. H2, Pd(OH)2/C, MeOH for target compound 5; b) 1. BF3·OEt2; 2. MeONa, MeOH; 3. PPh3, H2O; 4. RCOCl, aq. NaHCO3; 5. H2, Pd(OH)2/C, MeOH for target compounds 6–7; c) 0.5 mol eq 6-biotinamido hexanoic acid hydrazide, MeOH/H2O/AcOH 92![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :6:2, 40 °C, 24 h. :6:2, 40 °C, 24 h. | ||
Syntheses of target derivatives employed the coupling of appropriately tailored glycosyl trichloroacetimidate donors (8–12) with a common partially protected GlcNAc acceptor (13). Known trichloroacetimidates34 were prepared from five variously modified galactose derivatives: D-galactose (8, 97% yield), 6-deoxy-6-fluoro-D-galactose (9),35D-fucose (10)36 (6-deoxy-D-galactose), arabinose (11)37 (deshydroxymethyl-D-galactose) and 6-deoxy-6-azido-D-galactose (12).36 To access acceptor 13, glycosylation of a per-O-acetyl-β-GlcNAc with benzyl alcohol and ytterbium triflate,38,39 followed by deprotection under Zemplén conditions, gave the corresponding benzyl glycoside. A tert-butyldimethylsilyl protecting group was then introduced selectively on the primary hydroxyl to give the suitably protected glycosyl acceptor 13.40 Regioselective glycosylation41 of 13 catalysed by BF3 etherate gave β-1,4-linked disaccharides 14–18 containing the desired 6-modified galactose in 27–53% yield (Scheme 1).
Deprotection of compounds 14–17 by removal of the 6-O-silyl group, followed by de-O-acetylation under Zemplén conditions and removal of the anomeric benzyl protecting group by hydrogenolysis afforded target compounds 1–4. Azidodisaccharide 18 served as a vital divergent intermediate for amides 5–7. Staudinger reduction42 followed by per-O-acetylation and deprotection steps led to acetamide 5 from compound 18, whilst compounds 6 and 7 were accessed by Staudinger reduction and formation of amides under Schotten–Baumann conditions.43 In order to facilitate immobilisation of this disaccharide library on a neutravidin-coated SPR imaging chip, compounds 1–7 were chemically derivatised at their reducing ends with 6-biotinamido hexanoic acid hydrazide, giving biotinylated glycosyl hydrazides32,33 (1Bt–7Bt, Scheme 1) that were suitable for printing.
SPR imaging analysis of RCA120 binding to a 6-substituted galactoside library
In order to assess RCA120 recognition of unnatural galactoside derivatives, this modified library (1Bt–7Bt) was printed directly onto an SPR imaging chip, as described earlier. Equilibrium binding data are reported as a bar chart for glycans printed at 100 μg mL−1 concentration (ca. 150 μM) in Fig. 3.††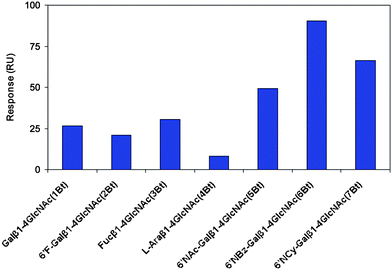 | ||
| Fig. 3 SPR imaging responses for equilibrium binding of RCA120 (800 nM) to an array of biotinylated Galβ1, 4-GlcNAc derivatives modified at the galactose 6-position (1Bt–7Bt). Data errors were typically in the ±10–15% range.13 | ||
As was immediately apparent from Fig. 3, replacement of the 6-hydroxyl group of galactose by fluorine (2Bt), or its removal altogether (3Bt), had limited impact on RCA120 binding (compared to 1Bt). In contrast, removal of the 5-hydroxymethyl group, to create 4Bt, essentially abolished binding. A significant increase (3–4 fold) in SPR responses was generated by the presence of the designed amides [6′-NHAc-(5Bt), 6′-NHBz-(6Bt) and 6′-NH.CO.C6H11-Galβ1-4GlcNAc (7Bt)), when compared to LacNAc (1Bt). These results are consistent with those from our initial screen of forty natural glycans, where 6′-sialyl LacNAc terminated glycans (notably Neu5Ac/Gc-α2-6Galβ1-4GlcNAc types; red) showed significant increased affinities. However, these unnatural ligands represent a dramatic further increase and are the most potent known ligands thus far developed, to the best of our knowledge.
STD NMR spectroscopy analysis of RCA120 binding to 6-modified galactosides
The data presented above clearly demonstrate that both terminal galactose and 6-modified galactose can act as motifs in ligands for RCA120. What these SPRi glycoarray data are unable to confirm is whether both classes of ligand bind at the same site on the lectin. We addressed this critical point through a solution assay based on STD NMR spectroscopy.44STD NMR relies on saturation transfer from the lectin to the bound fraction of ligand upon lectin irradiation at a selected resonance, followed by spin diffusion.45,46 This approach is finding increasing use in the analysis of carbohydrate–protein interactions.47,48 Upon addition of Galβ1-4Glc (1 mM) and the 6′-modified equivalent Neu5Acα2-6Galβ1-4Glc (1 mM) as ligands in excess to individual solutions of RCA120 (30 μM), the STD NMR spectra were recorded. Fig. 4A and 4B show the overlay of the 1D NMR reference spectra (in blue) and the corresponding STD NMR spectra (in red).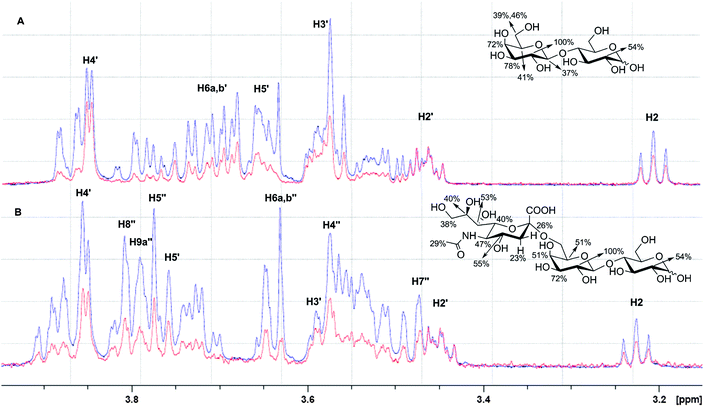 | ||
| Fig. 4 The interaction of (A) Galβ1-4Glc (Lac, 1 mM) and (B) Neu5Acα2-6Galβ1-4Glc (6′SL, 1 mM) with RCA120 (30 μM) by STD-NMR. The reference 1D NMR spectra (blue) are overlaid with the corresponding STD NMR spectra (red). The extent of saturation is expressed as relative intensity of the signals, normalised to the highest intensity signal (H2′-Gal for both (A) and (B)), to determine the binding epitopes. Gal residue protons are labelled as Hx′, whilst Neu5Ac protons are labelled as Hx′′. The chemical structures report the extent (%) of saturation for convenience. | ||
For Galβ1-4Glc (Fig. 4A), the saturation was transferred mainly onto H2′-, H3′- and H4′-Gal (100, 78 and 72%) and to a lesser extent to the other protons (H1′-Gal 37% [not shown in Fig], H5′-Gal 41% and H6a,b′-Gal 39 and 46%), consistent with literature data for methyl galactopyranoside.43 This suggested that H2′-, H3′- and H4′-Gal were in close contact with the RCA120 carbohydrate binding site. In the case of the 6′-modified equivalent Neu5Acα2-6Galβ1-4Glc (Fig. 4B), H2′- and H3′-Gal maintained the highest levels of saturation (100 and 72%, respectively), suggesting that these positions were integral to ligand recognition. Agreement with this observation can be found in lack of binding to RCA120 of α2,3Neu5Ac-terminating oligosaccharides (red bars, Fig. 1) - i.e. substitution of the 3-position of galactose precludes binding to RCA120, consistent with the 3 position of galactose being situated in close proximity to the protein on binding to RCA120. Significant differences were observed for saturation transfer for H4′-Gal signals in Galβ1-4Glc (72%) and Neu5Acα2-6Galβ1-4Glc (51%) and, to a lesser extent, for H5′-Gal (41% in Galβ1-4Glc; 51% in Neu5Acα2-6Galβ1-4Glc), reflecting changes in the network of interactions which stabilises the ligand into the binding sites, particularly around the 4 and 5 positions of galactose. The protons from the added 6′-NeuAc group (H4′′ to H9′′) were also subject to some saturation transfer (38–55%, Fig. 4B), suggesting proximity to the protein surface in the RCA120 complex.
Importantly, competition experiments using STD NMR with RCA120 in the presence of both Galβ1-4Glc and Neu5Acα2-6Galβ1-4Glc in various ratios (SI, Figs. S18–S21†) provided strong support for these two ligands adopting very similar binding modes and hence competing for the same binding site on RCA120.
Binding of 6-modified galactosides to RCA120 in other interaction studies
Despite the widespread notion that RCA120 is specific for non-reducing terminal β-linked D-galactose, numerous literature reports, employing a variety of techniques, give inconsistent evidence for the recognition of substituted galactosides. Early studies on glycan recognition reported that, α2-3Neu5Ac and α2-6Neu5Ac are “acceptable substituents on the terminal β-galactosyl moiety” for binding to ricin and its surrogate RCA120.49 However, while more recent observations by Gildersleeve and co-authors,50 using a fluorescence glycoarray approach, identified Neu5Acα2-6Gal structures as RCA120 binders (binding potency around 5 fold lower than lactose), α2-3Neu5Ac-containing glycans were not recognised in this assay. Data from frontal affinity chromatography studies using pyridylaminated (PA) oligosaccharides51 show that binding affinities of LacNAc-PA (KD 4.1 μM) and 6′-sialyl LacNAc-PA (KD 5.6 μM) to RCA120 were comparable. Conversely, studies carried out using ELISA reported very low, if any, binding of sialylated lactosides to RCA120.52 Fluorescence glycan array data from the Consortium for Functional Glycomics53 using polyacrylamide-based glycoconjugates54 show high RCA120 binding to non-reducing terminal Gal, Galβ1-4Glc and Galβ1-4GlcNAc. Interestingly, some but not all oligosaccharides terminating in the Neu5Acα2-6Galβ1-4GlcNAc sequence were also strongly recognised on these arrays (see SI, Fig. S17†), but glycans terminating in α2,3-Neu5Ac or α2,8-diNeu5Ac were not recognised. The SPRi data reported in the present study are in agreement with the Gildersleeve data, demonstrating binding to α2,6-Neu5Ac-Gal but not to α2,3-Neu5Ac-Gal (blue and red in Fig. 1, respectively). Can these conclusions be rationalised on a structural basis?Structural analysis of the binding of ricin and RCA120 with oligosaccharide ligands
The X-ray crystal structure of RCA120 bound to β-D-galactopyranose55 reveals that each molecule of the agglutinin incorporates one carbohydrate binding site capable of forming four hydrogen bonds with the β-Gal ligand: between Lys40 and OH-2, Asn46 and OH-3, Gly25 and OH-1, Asp22 and OH-4 (Fig. 5a). A hydrogen bonding network is further stabilised by hydrophobic interactions between Trp37 and the hydrophobic lower face of β-Gal (Fig. 5b). Overall, the crystallographic data clearly illustrate that the galactose 6-OH group does not make key polar contacts with the protein and suggest that substitution at this position may be accommodated; these observations are critically consistent with the data that we have determined in the present study. | ||
| Fig. 5 (a) Interaction of galactose (Gal) hydroxyl groups with amino acid (AA) residues of RCA120. In green, H-bond (Gal–OH⋯NH2–AA) distances expressed in Å (OH-2: 2.68, OH-3: 2.66, OH-4: 3.16); (b) In yellow, hydrophobic interactions between the lower face of Gal and Trp37 of RCA120 (PDB code 1RZO).55 | ||
This conclusion is further supported by low-energy conformer distribution analyses and nOe NMR studies56,57 on ricin and RCA120 using a series of deoxy-sugar derivatives of methyl β-lactoside. These studies have also highlighted the importance of positions 2, 3 and 4 in D-galactoside ligands as key polar groups for the interaction. Furthermore, STD NMR studies44 show saturation transfer from RCA120 to H-2, H-3 and H-4 (87, 100 and 87%, respectively) of methyl β-galactoside, but not to H-6, which is consistent with scope to accommodate modification of the galactose 6-OH group. Again, consideration of structural data supports the notion that alteration and substitution of the 6-position of galactose can be tolerated by RCA120.
Conclusions
The need for fast and sensitive detection methods for the unequivocal identification of ricin led us to explore SPR imaging of glycoarrays for screening ligands of the ricin surrogate RCA120. SPRi analysis of RCA120 interaction with a 40 compound glycoarray highlighted the potential of 6-modified D-galactosides to serve as ligand for this lectin; these observations were further substantiated by STD NMR spectroscopy. Non-natural 6-modified galactosides inspired by these observations were designed, synthesised and subsequently shown to exhibit up to a 3–4 fold enhanced RCA120 binding compared to the parent galactoside. To our knowledge, these are the most potent non-natural small molecule ligands currently known for RCA120. In summary, this study demonstrates the utility of glycoarrays and SPRi for both identifying and optimising carbohydrate ligands (Fig. 6) in the context of devising novel ligands for ricin sensor applications. | ||
| Fig. 6 RCA120 ligands explored and developed in this study. | ||
Experimental
Carbohydrate microarrays were prepared using biotinylated glycans, methods and hardware essentially as reported previously.12 Along with details of the synthesis, characterisation of the modified N-acetyllactosamines, protocols for the biotinylation of these reducing sugars and STD NMR competition experiments, this information can be found in the Electronic Supporting Information. All STD NMR experiments were performed on a Bruker Avance II 600 MHz spectrometer equipped with a TCI cryoprobe; spectra were processed on Bruker Topspin 2.1 software. A Bruker pulsed sequence for STD-NMR (Bruker pulse sequence code: stddiffesgp.3) was used with a shaped pulse train for saturation alternating between on and off resonance and a spoil pulse to destroy unwanted magnetization. The sequence includes solvent suppression using excitation sculpting with gradients and spinlock to suppress protein signals. On resonance was set at 360 Hz, whilst off resonance was set at −50,000 Hz. The number of scans varied with sample (typically 256). NMR samples were prepared in 500 μL of 99.9% D2O buffer containing 10 mM phosphate buffer, 150 mM NaCl at pH 7.4 (not corrected for D2O). Solute exchange was achieved by ultrafiltration of the RCA120 sample with Amicon® Ultra (Millipore) centrifugal filters (membrane cut-off 30 kDa). Addition of the saccharides Galβ1-4Glc and Neu5Acα2-6Galβ1-4Glc (Carbosynth Limited) to the protein NMR sample was from concentrated stock solutions in 99.9% buffered D2O. The ratio of protein to oligosaccharide was typically ca. 1:33. In competition experiments, the competing carbohydrate ligand was in 4-fold excess with respect to the primary carbohydrate ligand and therefore in a 1:132 ratio with the protein. As far as carbohydrate epitope mapping analysis was concerned, the STD integrals of the individual protons of the two disaccharides (Galβ1,4Glc and Neu5Acα2,6Galβ1,4Glc) were referenced to the strongest STD signal in each spectrum (H2′-Gal in both the saccharides), which was assigned a value of 100%.
Acknowledgements
These studies were supported by Research Councils UK Basic Technology Grant GR/S79268/02 (http://www.glycoarrays.org.uk/) and by the UK Engineering and Physical Sciences Research Council Grant EP/G037604/1. Biotinylated carbohydrate derivatives used in this study were kindly provided by the Carbohydrate Synthesis Core of the Consortium for Functional Glycomics (http://www.functionalglycomics.org/fg/), which is funded by US National Institute of General Medical Sciences grant GM62116. BGD is a Royal Society Wolfson Merit Award recipient.References
- L. L. Kiessling and R. A. Splain, Annu. Rev. Biochem., 2010, 79, 619–653 CrossRef CAS.
- K. Larsen, M. B. Thygesen, F. Guillaumie, W. G. T. Willats and K. J. Jensen, Carbohydr. Res., 2006, 341, 1209–1234 CrossRef CAS.
- D. M. Ratner, E. W. Adams, M. D. Disney and P. H. Seeberger, ChemBioChem, 2004, 5, 1375–1383 CrossRef CAS.
- J. E. Turnbull and R. A. Field, Nat. Chem. Biol., 2007, 3, 74–77 CrossRef CAS.
- M. Fais, R. Karamanska, D. A. Russell and R. A. Field, J. Cereal Sci., 2009, 50, 306–311 CrossRef CAS.
- L. Krishnamoorthy and L. K. Mahal, ACS Chem. Biol., 2009, 4, 715–732 CrossRef CAS.
- O. Oyelaran and J. C. Gildersleeve, Curr. Opin. Chem. Biol., 2009, 13, 406–413 CrossRef CAS.
- Y. Liu, A. S. Palma and T. Feizi, Biol. Chem., 2009, 390, 647–656 CrossRef CAS.
- N. Laurent, J. Voglmeir and S. L. Flitsch, Chem. Commun., 2008, 4400–4412 RSC.
- X. Chen and A. Varki, ACS Chem. Biol., 2010, 5, 163–176 CrossRef CAS.
- O. Blixt, S. F. Han, L. Liao, Y. Zeng, J. Hoffmann, S. Futakawa and J. C. Paulson, J. Am. Chem. Soc., 2008, 130, 6680–6681 CrossRef CAS.
- C. L. Schofield, B. Mukhopadhyay, S. M. Hardy, M. B. McDonnell, R. A. Field and D. A. Russell, Analyst, 2008, 133, 626–634 RSC.
- R. Karamanska, J. Clarke, O. Blixt, J. I. Macrae, J. Q. Q. Zhang, P. R. Crocker, N. Laurent, A. Wright, S. L. Flitsch, D. A. Russell and R. A. Field, Glycoconjugate J., 2008, 25, 69–74 CrossRef CAS.
- J. Audi, M. Belson, M. Patel, J. Schier and J. Osterloh, JAMA, J. Am. Med. Assoc., 2005, 294, 2342–2351 CrossRef CAS.
- F. Musshoff and B. Madea, Drug Test. Anal., 2009, 1, 184–191 Search PubMed.
- B. Stechmann, S. K. Bai, E. Gobbo, R. Lopez, G. Merer, S. Pinchard, L. Panigai, D. Tenza, G. Raposo, B. Beaumelle, D. Sauvaire, D. Gillet, L. Johannes and J. Barbier, Cell, 2010, 141, 231–242 CrossRef CAS.
- R. R. Kale, C. M. McGannon, C. Fuller-Schaefer, D. M. Hatch, M. J. Flagler, S. D. Gamage, A. A. Weiss and S. S. Iyer, Angew. Chem., Int. Ed., 2008, 47, 1265–1268 CrossRef CAS.
- H. Uzawa, K. Ohga, Y. Shinozaki, I. Ohsawa, T. Nagatsuka, Y. Seto and Y. Nishida, Biosens. Bioelectron., 2008, 24, 923–927 CrossRef CAS.
- S. Takae, Y. Akiyama, H. Otsuka, T. Nakamura, Y. Nagasaki and K. Kataoka, Biomacromolecules, 2005, 6, 818–824 CrossRef CAS.
- N. P. Pera, A. Kouki, S. Haataja, H. M. Branderhorst, R. M. J. Liskamp, G. M. Visser, J. Finne and R. Pieters, Org. Biomol. Chem., 2010, 8, 2425–2429 RSC.
- B. Mukhopadhyay, M. B. Martins, R. Karamanska, D. A. Russell and R. A. Field, Tetrahedron Lett., 2009, 50, 886–889 CrossRef CAS.
- K. A. Barth, G. Coullerez, L. M. Nilsson, R. Castelli, P. H. Seeberger, V. Vogel and M. Textor, Adv. Funct. Mater., 2008, 18, 1459–1469 CrossRef CAS.
- K. El-Boubbou, C. Gruden and X. Huang, J. Am. Chem. Soc., 2007, 129, 13392–13393 CrossRef CAS.
- O. J. Barrett, J. L. Childs and M. D. Disney, ChemBioChem, 2006, 7, 1882–1885 CrossRef CAS.
- M. D. Disney and P. H. Seeberger, Chem. Biol., 2004, 11, 1701–1707 CrossRef CAS.
- R. Hegde and S. K. Podder, Eur. J. Biochem., 1998, 254, 596–601 CrossRef CAS.
- B. Liu, Z. Y. Tong, Y. H. Tian, L. Q. Hao and X. H. Mu, Chin. J. Anal. Chem., 2006, 34, 1779–1782 CAS.
- A. Varki; R. Cummings; J. D. Esko; H. H. Freeze; P. Stanley; C. Bertozzi; G. W. Hart; M. E. Etzler, in Essentials of Glycobiology, Cold Spring Harbor Laboratory Press, New York, 2009 Search PubMed.
- (a) V. Kodoyianni, BioTechniques, 2011, 50, 32–40 CrossRef CAS; (b) S. Ray, G. Mehta and S. Srivastava, Proteomics, 2010, 10, 731–748 CrossRef CAS; (c) S. Scarano, M. Mascini, A. P. F. Turner and M. Minunni, Biosens. Bioelectron., 2010, 25, 957–966 CrossRef CAS.
- S. H. Millen, D. M. Lewallen, A. B. Herr, S. S. Iyer and A. A. Weiss, Biochemistry, 2010, 49, 5954–5967 CrossRef CAS.
- R. L. Rich and D. G. Myszka, J. Mol. Recognit., 2010, 23, 1–64 CAS.
- Y. Shinohara, H. Sota, F. Kim, M. Shimizu, M. Gotoh, M. Tosu and Y. Hasegawa, J. Biochem., 1995, 117, 1076–1082 CAS.
- C. Leteux, R. A. Childs, W. G. Chai, M. S. Stoll, H. Kogelberg and T. Feizi, Glycobiology, 1998, 8, 227–236 CrossRef CAS.
- R. R. Schmidt and W. Kinzy, Adv. Carbohydr. Chem. Biochem., 1994, 50, 21–123 CAS.
- W. Stahl, U. Sprengard, G. Kretzschmar and H. Kunz, Angew. Chem., Int. Ed. Engl., 1994, 33, 2096–2098 CrossRef.
- C. W. T. Chang, Y. Hui, B. Elchert, J. H. Wang, J. Li and R. Rai, Org. Lett., 2002, 4, 4603–4606 CrossRef CAS.
- T. Murakami, H. Matsuda, M. Inadzuki, K. Hirano and M. Yoshikawa, Chem. Pharm. Bull., 1999, 47, 1717–1724 CAS.
- C. F. Crasto and G. B. Jones, Tetrahedron Lett., 2004, 45, 4891–4894 CrossRef CAS.
- S. A. Allman, H. H. Jensen, B. Vijayakrishnan, J. A. Garnett, E. Leon, Y. Liu, D. C. Anthony, N. R. Sibson, T. Feizi, S. Matthews and B. G. Davis, ChemBioChem, 2009, 10, 2522–2529 CrossRef CAS.
- S. Adam, Tetrahedron Lett., 1988, 29, 6589–6592 CrossRef CAS.
- Z. H. Gan, S. D. Cao, Q. Q. Wu and R. Roy, J. Carbohydr. Chem., 1999, 18, 755–773 CrossRef CAS.
- A. Bianchi and A. Bernardi, Tetrahedron Lett., 2004, 45, 2231–2234 CrossRef CAS.
- P. Boullanger, V. Maunier and D. Lafont, Carbohydr. Res., 2000, 324, 97–106 CrossRef CAS.
- M. Mayer and B. Meyer, J. Am. Chem. Soc., 2001, 123, 6108–6117 CrossRef CAS.
- K. Lycknert, M. Edblad, A. Imberty and G. Widmalm, Biochemistry, 2004, 43, 9647–9654 CrossRef CAS.
- F. J. Munoz, J. I. Santos, A. Arda, S. Andre, H. J. Gabius, J. V. Sinisterra, J. Jimenez-Barbero and M. Hernaiz, Org. Biomol. Chem., 2010, 8, 2986–2992 RSC.
- C. Rademacher, J. Landstrom, N. Sindhuwinata, M. M. Palcic, G. Widmalm and T. Peters, Glycoconjugate J., 2010, 27, 349–358 CrossRef CAS.
- M. R. Richards and T. L. Lowary, ChemBioChem, 2009, 10, 1920–1938 CrossRef CAS.
- I. E. Liener; N. Sharon and I. J. Goldstein, in The Lectins: Properties, Functions, and Applications in Biology and Medicine, Academic Press, New York, 1986; pp. 180–185 Search PubMed.
- J. C. Manimala, T. A. Roach, Z. T. Li and J. C. Gildersleeve, Angew. Chem., Int. Ed., 2006, 45, 3607–3610 CrossRef CAS.
- Y. Itakura, S. Nakamura-Tsuruta, J. Kominami, N. Sharon, K. Kasai and J. Hirabayashi, J. Biochem., 2007, 142, 459–469 CrossRef CAS.
- A. M. Wu, J. H. Wu, T. Singh, L. J. Lai, Z. G. Yang and A. Herp, Mol. Immunol., 2006, 43, 1700–1715 CrossRef CAS.
- http://www.functionalglycomics.org/static/index.shtml .
- N. V. Bovin, Glycoconjugate J., 1998, 15, 431–446 CrossRef CAS.
- N. V. Konareva, A. G. Gabdulkhakov, S. Eschenburg, S. Stoeva, A. N. Popov, R. Krauspenhaar, M. E. Andrianova, Y. Savochkina, I. I. Agapov, A. G. Tonevitskii, A. N. Kornev, V. V. Kornilov, V. N. Zaitsev, W. Voelter, C. Betzel, S. V. Nikonov, B. K. Vainshtein and A. M. Mikhailov, Crystallogr. Rep., 2001, 46, 792–800 CrossRef.
- A. Rivera-Sagredo, D. Solis, T. Diaz Maurino, J. Jimenez-Barbero and M. Martin-Lomas, Eur. J. Biochem., 1991, 197, 217–228.
- D. Solis, P. Fernandez, T. Diaz Maurino, J. Jimenez Barbero and M. Martin Lomas, Eur. J. Biochem., 1993, 214, 677–683 CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c1sc00120e |
| ‡ These authors contributed equally to this work. |
| § Present address: Department of Chemistry, University of Canterbury, Private Bag 4800, Christchurch 8140, New Zealand. |
| ¶ Current analytical procedures for the detection of ricin are summarised in ref. 15. |
| || Abbreviations: Gal (galactose), GalNAc (N-acetylgalactosamine), Glc (glucose), GlcNAc (N-acetylglucosamine), NeuAc-2,3 (α2,3-linked N-acetylneuraminic acid), NeuGc-2,3 (α-2,3-linked N-glycolylneuraminic acid), NeuAc-2,6 (α-2,6-linked N-acetylneuraminic acid), NeuGc-2,6 (α-2,6-linked N-glycolylneuraminic acid) and NeuAc-2,8 (α-2,8-linked N-acetylneuraminic acid). Colloquially, N-acetylneuraminic acid is often simply referred to as sialic acid. |
| ** The relative ranking of binding events are used throughout this study. Consistent with problems experienced elsewhere for quantifying protein–carbohydrate interactions by SPR,30 and cautionary notes on the subject, direct fitting of SPR data to simple 1:1 or 2:1 binding models is not always possible, or appropriate.31 |
| †† SPR signals obtained for printed glycosyl hydrazones interacting with lectins were routinely much lower than for the corresponding glycosides (compare Fig. 5 and Fig. 2, respectively). However, where investigated, the relative ranking of binding was independent of the nature of the linkage. |
| This journal is © The Royal Society of Chemistry 2011 |