Redox couple related influences of π-conjugation extension in organic dye-sensitized mesoscopic solar cells†
Min
Zhang
,
Jingyuan
Liu
,
Yinghui
Wang
,
Difei
Zhou
and
Peng
Wang
*
State Key Laboratory of Polymer Physics and Chemistry, Changchun Institute of Applied Chemistry, Chinese Academy of Sciences, Changchun, 130022, China. E-mail: peng.wang@ciac.jl.cn.; Fax: +86 431 85262953; Tel: +86 431 85262953
First published on 17th May 2011
Abstract
The iodide/triiodide redox shuttle is an unparalleled option in making efficient dye-sensitized solar cells (DSCs) but also presents some prominent constraints on the development of new photosensitizers, semiconductors and counter electrodes. In this paper, a cobalt electrolyte has been identified to offer an advantageous impact of π-conjugation extension in push–pull organic dyes, upon the interfacial charge recombination kinetics and thus the open-circuit photovoltage of DSCs, in sharp contrast with the conventional iodine congener. The usage of a solely visible-light-absorption dye has generated a DSC free of corrosive iodine, exhibiting an impressive overall power conversion efficiency of 8.0% at the 100 mW cm−2 AM1.5G conditions.
Introduction
Dye-sensitized solar cells (DSCs) are the theme of intense research owing to their grand potential to cut the cost of solar electricity. In past years, a mass of endeavours have been devoted to the synthesis of a great variety of push–pull organic photosensitizers characteristic of high absorption coefficients.1 To elaborately tailor metal-free chromophores which can harness visible as well as near-infrared solar photons, the elongation of π-conjugated spacers is one common tactic of choice apart from the judicious selection of electron donors and acceptors. The incorporation of a longer conjugated segment in a push–pull dye does victoriously red-shift the photocurrent response of DSCs with iodide/triiodide as the redox shuttle of electrolytes, but exclusively brings forth a reduction of cell photovoltage.2 The general latter observation considerably chills the enthusiasm for further design of near-infrared-absorption dyestuffs that are highly desirable for more efficient DSCs.Our recent experiments have shown that in prominent contrast to the traditional iodine electrolyte, a tris(1,10-phenanthroline)cobalt counterpart can endow dye-sensitized solar cells with a perennially yearning feature, i.e. an increment of the cell photovoltage concomitant with an extension of the π-conjugated linker in organic dyes. In this paper, we employ two simple model photosensitizers T12i and T32k (Fig. 1A) to demonstrate the redox couple dependent trait of metal-free dyes in DSCs, and further take a close look at its intrinsic origins through a joint electrical, photophysical, and computational analysis.
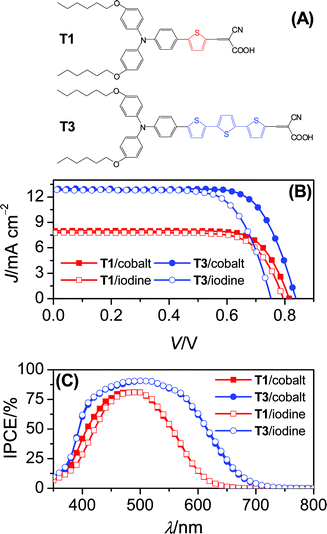 | ||
| Fig. 1 (A) Dye structures, (B) J–V characteristics under irradiation of 100 mW cm−2 simulated AM1.5G sunlight and (C) photocurrent action spectra. Cell area tested with a metal mask: 0.158 cm2. An anti-reflection film was adhered to the testing cell during measurements. | ||
Results and discussion
Our cobalt electrolyte is composed of 0.3 M tris(1,10-phenanthroline)cobalt(II) di[bis(trifluoromethanesulfonyl)imide],3 0.1 M nitrosonium tetrafluoroborate, 0.5 M 4-tert-butylpyridine (TBP) and 0.1 M lithium bis(trifluoromethanesulfonyl)imide (LiTFSI) in acetonitrile. A control electrolyte based on the conventional iodine redox mediator was also formulated and its composition is as follows: 0.3 M 1,3-dimethylimidazolium iodide,4 0.1 M iodine, 0.5 M TBP and 0.1 M LiTFSI in acetonitrile. The details of cell fabrication are presented in the experimental section. Current–voltage (J–V) characteristics (Fig. 1B) under irradiation of 100 mW cm−2 simulated AM1.5G sunlight were first recorded and the detailed cell parameters are collected in Table 1. In general, the π-conjugation extension from T1 to T3 offers a comparable increment of short-circuit photocurrent density (Jsc), regardless of the redox couple selection of either cobalt or iodine. For cells with the cobalt electrolyte, it is very encouraging to note that an increase of the thiophene unit from 1 to 3 leads to a 22 mV augmentation of open-circuit photovoltage (Voc) from 815 to 837 mV, which is dramatically contrasted by an opposite Voc variation tendency of the iodine cells (T1: 795 mV; T3: 752 mV). Moreover, the T3cell with the cobalt electrolyte displays an impressive overall conversion efficiency of 8.0% amongst diverse iodine-free DSCs reported previously.3,5| Dye/electrolyte | T1/cobalt | T3/cobalt | T1/iodine | T3/iodine |
|---|---|---|---|---|
| a The spectral distribution of our light resource simulates the AM1.5G solar emission (ASTM G173-03). The validity of our data is further confirmed by comparing the overlap integral of the measured IPCE spectrum with the standard AM1.5G emission spectrum and the experimental Jsc, showing a less than 5% error. | ||||
| J sc (mA cm−2) | 7.99 | 12.98 | 7.78 | 12.86 |
| V oc (mV) | 815 | 837 | 795 | 752 |
| FF | 0.76 | 0.74 | 0.75 | 0.71 |
| η (%) | 5.0 | 8.0 | 4.6 | 6.9 |
| E c–EF,redox (eV) | 1.11 | 1.12 | 1.03 | 1.05 |
| β | 0.72 | 0.65 | 0.64 | 0.77 |
| U 0k (cm−3s−1) | 3.5 × 1023 | 1.2 × 1023 | 2.3 × 1021 | 8.4 × 1022 |
The open-circuit photovoltage of a DSC is dictated by the titania conduction band edge (Ec) and electrolyte Fermi-level (EF,redox) as well as the photocurrent generation and interfacial charge recombination rates. To examine the possible impact of π-conjugation extension on Ec, we carried out electrical impedance spectroscopy (EIS) measurements6 to first derive the chemical capacitance (Cμ) at the titania/dye/electrolyte interface, which can be described as7
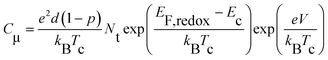 | (1) |
 | ||
| Fig. 2 (A) Chemical capacitance (Cμ) and (B) interfacial charge transfer resistance (Rct) plotted as a function of potential bias (V). | ||
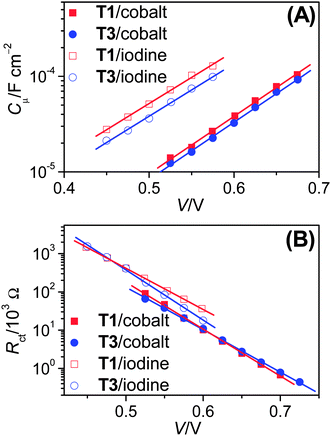
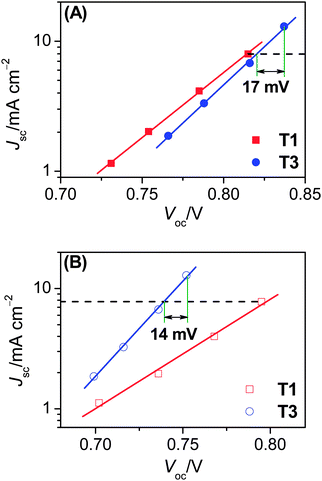 | ||
| Fig. 3 Plots of short-circuit photocurrent density (Jsc) versus open-circuit photovoltage (Voc) for cells with the (A) cobalt and (B) iodine electrolytes under different illuminations. | ||
On the basis of above analysis, we may deduce that the variation of charge recombination kinetics along with the photosensitizer alteration from T1 to T3 could be in distinctive fashions for the cobalt and iodine electrolytes. The rate of interfacial charge recombination (Un) with cobalt(III) or triiodide can be described through the following expression based on the nonlinear recombination model:8,9
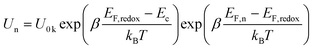 | (2) |
Here EF,n is the titania Fermi-level, T is the absolute temperature and U0k serves as an effective recombination rate constant with U0k = k0Ncβ, where k0 is the apparent rate constant, β is the reaction order of titania electrons and Nc is the density of accessible states in the titania conduction band. Furthermore, modelling EIS can also afford the charge-transfer resistance Rct as a function of potential bias V (Fig. 2B), from which β (Table 1) and k0 can be fitted through10
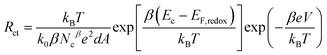 | (3) |
To gain insight into the mechanistic basis behind the highly redox couple dependent influence of π-conjugation extension upon interfacial charge recombination, we further performed quantum calculation to scrutinize the possible interactions of dye molecules with electron-accepting species in electrolytes. In view of the highly polarizable nature of iodine, the employment of the iodine redox couple may lead to the formation of dye-I2 and/or dye-I3− complexes.2h,11 The calculated dipole moments (μD) and polarizabilities of these two photosensitizers are listed in Table S1.† With respect to the T1dye, T3 features a larger μD value and an evidently increased polarizability (T1: 87.5 Å3; T3: 130.3 Å3), which could induce a stronger intermolecular interaction of dye molecules with I2 and/or I3−. Moreover, our quantum calculations have also uncovered that the T3chromophore with more active sulphur sites is more prone to form dye-iodine complexes (Fig. 4), featuring some larger interaction energies between iodine and sulphur. Thereby, it can be derived that the conjugation length correlated dye/iodine interaction evokes a higher iodine concentration in the vicinity of titania anchored with the T3dye, rationalizing its faster interfacial charge recombination kinetics. On the other side, compared to that of the iodine counterpart, a larger steric hindrance of the bulky cobalt(III) complex could prevent its close approach to the surface of titania. Furthermore, a longer π-conjugated spacer probably increases the electron tunnelling distance between titania and cobalt(III), leading to a relatively slower interception of photoinjected electrons by cobalt(III).
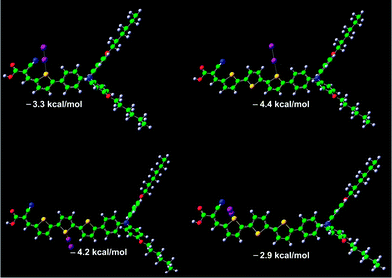 | ||
| Fig. 4 Optimized geometries of T1-I2 and T3-I2 complexes. The interaction energies between iodine and sulfur are also included. Iodine: purple; carbon: green; sulfur: yellow; nitrogen: blue; oxygen: red; hydrogen: gray. | ||
As Fig. 1C presents, the incident photon-to-collected electron conversion efficiencies (IPCEs) of cells based on the cobalt or iodine electrolyte are plotted as a function of wavelength. Both electrolytes confer a comparable IPCE summit on the T1 or T3 coated titania film, while a dye dependent maximum is indeed perceived, which can be understood in terms of a low absorption coefficient of T1 as compared to T3 (Fig. S2†). Additionally, the cobalt electrolyte endows the T1cell with evidently higher photocurrent response from 375 to 480 nm than the iodine congener. This is caused by a dissipative light absorption of the iodine electrolyte, which competes with the weak absorption of a T1-coated titania film in the blue region.
Laser-induced transient absorption traces were further measured to probe the kinetics of oxidized dye related charge transfer reactions, i.e. its hole transfer either to titania or electron-donating species in electrolytes, allowing one to evaluate the contribution of this dual-channel kinetic competition to the IPCE height of a DSC.12 Prior to this, we first recorded the transient absorption difference spectra (Fig. 5A) of dye-coated titania films with an ICCD camera, from which a strong absorption in the near-infrared region can be identified for both T1 and T3dye cations, with respect to their neutral counterparts. On account of the signal sensitivity of our PMT detector, a monochromatic probe light at 782 nm was chosen in the succedent kinetic measurements, where the excitation wavelength and fluence were carefully controlled to ensure that ∼2.0 × 1013photons were absorbed by a dye-coated titania film (∼0.8 photon per nanoparticle), with an identical exciton distribution profile during every laser pulse.
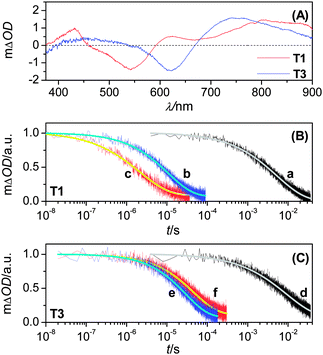 | ||
| Fig. 5 (A) Transient absorption spectra upon laser excitation at 500 nm of 2.8-μm-thick, dye-coated titania films immersed in an inert electrolyte composed of 0.1 M LiTFSI and 0.5 M TBP in acetonitrile. (B, C) Transient absorption kinetics of 10.0 μm-thick, dye-coated titania films immersed in the inert (traces a and d), cobalt (traces b and e) or iodine (traces c and f) electrolyte. Excitation wavelength and pulse fluence: 51 μJ cm−2 at 580 nm (traces a, b, and c); 46 μJ cm−2 at 640 nm (traces d, e, and f). The smooth lines are stretched exponential fittings over raw data obtained by averaging over 500 laser shots. | ||
In the presence of an inert electrolyte consisting of 0.1 M LiTFSI and 0.5 M TBP in acetonitrile, the slow absorption decays (traces a and d in Fig. 5B and 5C) can be assigned to the charge recombination reaction between dye cations and titania electrons. Upon utilizing the realistic electrolytes for cell fabrication, however, the absorption decays (traces b, c, e and f) are notably accelerated owing to occurrence of dye regeneration by cobalt(II) or iodide. The half-reaction times (t1/2) of these charge transfer processes can be derived through fitting with a stretched exponential function and are collected in Table S2.† Evidently, both π-conjugation extension and redox couple alteration can result in a profound influence on dye regeneration kinetics. However, the kinetic branch ratios of these two hole capture reactions by electrolytes and titania electrons exceed 160 in all of our four cells, manifesting that almost 100% of the dye cations can be intercepted by the iodide or cobalt(II) ions. A ∼60 mV negative shift of the formal potential from T1 to T3 (Fig. S3†) generally brings on a deceleration of dye regeneration in both cobalt and iodine electrolytes, which however displays a substantially distinctive dependence on the redox couple selection (cobalt: ∼3-fold deceleration; iodine: ∼18-fold deceleration). This important observation indicates that with respect to the routinely used iodine electrolyte, the cobalt counterpart should allow for a large uplifting space of ground-state redox potential in the further design of low energy gap push–pull dyes to harvest the near-infrared solar photons.
Conclusions
To summarize, by use of a cobalt redox electrolyte we have demonstrated a favourable impact of the π-conjugation extension of organic photosensitizers upon the photovoltage of dye-sensitized solar cells for the first time, in striking contrast to the previously general observation on DSCs based on an iodine electrolyte. The usage of a solely visible-light-absorption dye has generated a DSC free of corrosive iodine, exhibiting an impressive overall power conversion efficiency of 8.0% under 100 mW cm−2 AM1.5G conditions. We anticipate that a remarkable efficiency enhancement of iodine-free DSCs could be achieved in the near future, through sensible exploitation of narrow energy gap photosensitizers and energy-level engineering of redox shuttles.Experimental section
Cell fabrication
A bilayer titania film was employed as the negative electrode of DSCs. On a precleaned FTO conducting glass electrode (NSG, Solar, 4 mm thick), a 2.8 μm-thick transparent layer (porosity: 0.64) consisting of 23 nm-sized titania particles was first screen-printed and further coated by a 4.1 μm-thick second layer of scattering titania particles (WER2-O, Dyesol). The film thickness was examined with a bench-top Ambios XP-1 stylus profilometer. The preparation procedures of TiO2 nanocrystals, paste for screen-printing and nanostructured TiO2 film were reported in a previous paper.13 A cycloidal TiO2 electrode (∼0.28 cm2) was stained by immersing it into a tetrahydrofuran solution containing 150 μM dye and 30 mM 3α,7α-dihydroxy-5β-cholic acid coadsorbent overnight. After rinsing with acetonitrile and drying by air flow, the dye-coated titania electrode was assembled with a thermally platinized FTO positive electrode possessing an electrolyte-perfusion hole, which was beforehand produced with a sand-blasting drill. The two electrodes were separated by a 30 μm-thick Bynel (DuPont) hot-melt gasket. The internal space was perfused with a liquid electrolyte with a vacuum back filling system. Ultimately, the hole on the positive electrode was closed hermetically with a Bynel sheet and a thin glass cover by heating.Photovoltaic characterizations
A Keithley 2400 source meter and a Zolix Omni-λ300 monochromator equipped with a 500 W xenon lamp were used for photocurrent action spectrum measurements, with a wavelength sampling interval of 10 nm and a current sampling time of 2 s under the full computer control. A Hamamatsu S1337–1010BQ silicon diode used for IPCE measurements was calibrated in National Institute of Metrology, China. A model LS1000-4S-AM1.5G-1000W solar simulator (Solar Light Company, USA) in combination with a metal mesh was employed to give an irradiance of 100 mW cm−2. The light intensity was tested with a PMA2144 pyranometer and a calibrated PMA 2100 dose control system. Current–voltage (J–V) characteristics were obtained by applying a bias potential to a testing cell and measuring photocurrent with a Keithley 2400 source meter under the full computer control. The measurements were fully automated using Labview 8.0. A metal mask with an aperture area of 0.158 cm2 was covered on a testing cell during all measurements. An anti-reflection film (λ < 380 nm, ARKTOP, ASAHI Glass) was adhered to the DSC photoanode during IPCE and J–V measurements. The short-circuit photocurrent densities measured under this solar simulator are consistent with the integral of IPCEs over the AM1.5G spectrum (ASTM G173-03), within a 5% error.Voltammetry measurements
We first constructed two reference electrodes composed of platinum wires dipped in the DSC electrolyte-filling glass tubes, one end of which was pre-sealed with a porous ceramic frit, to measure the square-wave voltammograms of ferrocene dissolved in acetonitrile with a CHI660C electrochemical workstation. A 5 μm-radius platinum ultramicroelectrode was used as working electrode and a platinum foil as auxiliary electrode. On the basis of voltammetric measurements, we further calculated EF,redox of the cobalt and iodine electrolytes being −5.110 and −4.885 eV versus vacuum, respectively. Cyclic voltammetric measurements of dye-coated titania films on fluorine-doped tin oxide (FTO) were performed with a platinum gauze counter electrode and an Ag/AgCl (sat. KCl) auxiliary electrode. Redox potentials and electrolyte Fermi-levels reported in this paper were calibrated with ferrocene as the reference (−5.14 V versus vacuum).Electrical impedance measurements
We performed electrical impedance measurements under illumination of a white light-emitting diode with an IM6ex electrochemical workstation. We selected a frequency range from 50 mHz to 100 kHz and a potential modulation of 20 mV. To meet the zero current condition, a potential bias (V) being equal to the open-circuit photovoltage (Voc) was applied to the illuminated cell. The measured impedance spectra were further fitted using the Z-view software (v2.80, Scribner Associates Inc.).Steady-state and transient absorption measurements
Steady-state electronic absorption spectra were recorded on an Agilent G1103A spectrometer. Transient absorption measurements were performed on a LP920 laser flash spectrometer in conjunction with a nanosecond tunable OPOLett 355II laser. Transient absorption spectra were measured with an Andor iStar ICCD camera, and the absorption kinetic traces were detected by a fast photomultiplier tube and recorded with a TDS 3012C digital oscillograph.Computational details
The ground-state geometries were first optimized by the density functional theory (DFT)14 method, with hybrid Beck's three-parameter functional and Lee–Yang–Parr functional (B3LYP).15,16 Dipole moments and polarizabilities were calculated at the B3LYP level based on the optimized geometries. The LANL2DZ basis set was used for the iodine atom and 6-31G(d,p) basis set for other atoms. All calculations reported here were performed with the Gaussian09 program package.17Acknowledgements
The National 973 Program (2007CB936702 and 2011CBA00702), the National Science Foundation of China (50973105), the CAS Knowledge Innovation Program (KGCX2-YW-326), and the CAS Hundred Talent Program are acknowledged for financial support.Notes and references
- For a comprehensive review, see: A. Mishra, M. K. R. Fischer and P. Bauerle, Angew. Chem., Int. Ed., 2009, 48, 2474 Search PubMed.
- Selected examples include (a) K. Hara, M. Kurashige, Y. Dan-oh, C. Kasada, A. Shinpo, S. Suga, K. Sayama and H. Arakawa, New J. Chem., 2003, 27, 783 RSC; (b) T. Kitamura, M. Ikeda, K. Shigaki, T. Inoue, N. A. Anderson, X. Ai, T. Lian and S. Yanagida, Chem. Mater., 2004, 16, 1806 CrossRef CAS; (c) K. R. J. Thomas, J. T. Lin, Y.-C. Hsu and K.-C. Ho, Chem. Commun., 2005, 4098 RSC; (d) S. Kim, J. K. Lee, S. O. Kang, J. Ko, J.-H. Yum, S. Fantacci, F. De Angelis, D. Di Censo, M. K. Nazeeruddin and M. Grätzel, J. Am. Chem. Soc., 2006, 128, 16701 CrossRef CAS; (e) D. P. Hagberg, T. Marinado, K. M. Karlsson, K. Nonomura, P. Qin, G. Boschloo, T. Brinck, A. Hagfeldt and L. Sun, J. Org. Chem., 2007, 72, 9550 CrossRef CAS; (f) R. Chen, X. Yang, H. Tian, X. Wang, A. Hagfeldt and L. Sun, Chem. Mater., 2007, 19, 4007 CrossRef CAS; (g) K. R. J. Thomas, Y.-C. Hsu, J. T. Lin, K.-M. Lee, K.-C. Ho, C.-H. Lai, Y.-M. Cheng and P.-T. Chou, Chem. Mater., 2008, 20, 1830 CrossRef CAS; (h) M. Miyashita, K. Sunahara, T. Nishikawa, Y. Uemura, N. Koumura, K. Hara, A. Mori, T. Abe, E. Suzuki and S. Mori, J. Am. Chem. Soc., 2008, 130, 17874 CrossRef CAS; (i) R. Li, X. Lv, D. Shi, D. Zhou, Y. Cheng, G. Zhang and P. Wang, J. Phys. Chem. C, 2009, 113, 7469 CrossRef CAS; (j) J. T. Lin, P.-C. Chen, Y.-S. Yen, Y.-C. Hsu, H.-H. Chou and M.-C. P. Yeh, Org. Lett., 2009, 11, 97 CrossRef CAS; (k) J. Liu, R. Li, X. Si, D. Zhou, Y. Shi, Y. Wang, X. Jing and P. Wang, Energy Environ. Sci., 2010, 3, 1924 RSC.
- H. Nusbaumer, S. M. Zakeeruddin, J.-E. Moser and M. Grätzel, Chem.–Eur. J., 2003, 9, 3756 CrossRef CAS.
- P. Bonhôte, A.-P. Dial, N. Papageorgiou, K. Kalyanasundaram and M. Grätzel, Inorg. Chem., 1996, 35, 1168 CrossRef CAS.
- For a comprehensive review, see: (a) S. Yanagida, Y. Yu and K. Manseki, Acc. Chem. Res., 2009, 42, 1827 CrossRef CAS; (b) G. Oskam, B. V. Bergeron, G. J. Meyer and P. C. Searson, J. Phys. Chem. B, 2001, 105, 6867 CrossRef CAS; (c) S. A. Sapp, C. M. Elliott, C. Contado, S. Caramori and C. A. Bignozzi, J. Am. Chem. Soc., 2002, 124, 11215 CrossRef CAS; (d) P. Wang, S. M. Zakeeruddin, J.-E. Moser, R. Humphry-Baker and M. Grätzel, J. Am. Chem. Soc., 2004, 126, 7164 CrossRef CAS; (e) S. Hattori, Y. Wada, S. Yanagida and S. Fukuzumi, J. Am. Chem. Soc., 2005, 127, 9648 CrossRef CAS; (f) Z. Zhang, P. Chen, T. N. Murakami, S. M. Zakeeruddin and M. Grätzel, Adv. Funct. Mater., 2008, 18, 341 CrossRef CAS; (g) M. Wang, N. Chamberland, L. Breau, J.-E. Moser, R. Humphry-Baker, B. Marsan, S. M. Zakeeruddin and M. Grätzel, Nat. Chem., 2010, 2, 385 CrossRef CAS; (h) S. M. Feldt, E. A. Gibson, E. Gabrielsson, L. Sun, G. Boschloo and A. Hagfeldt, J. Am. Chem. Soc., 2010, 132, 16714 CrossRef CAS; (i) T. C. Li, A. M. Spokoyny, C. She, O. K. Farha, C. A. Mirkin, T. J. Marks and J. T. Hupp, J. Am. Chem. Soc., 2010, 132, 4580 CrossRef CAS; (j) T. Daeneke, T.-H. Kwon, A. B. Holmes, N. W. Duffy, U. Bach and L. Spiccia, Nat. Chem., 2011, 3, 211 CAS; (k) Y. Bai, Q. Yu, N. Cai, Y. Wang, M. Zhang and P. Wang, Chem. Commun., 2011, 47, 4376 RSC; (l) D. Zhou, Q. Yu, N. Cai, Y. Bai, Y. Wang and P. Wang, Energy Environ. Sci., 2011, 4 10.1039/C0EE00735H.
- J. Bisquert, J. Phys. Chem. B, 2002, 106, 325 CrossRef CAS.
- E. M. Barea, J. Ortiz, F. J. Payá, F. Fernández-Lázaro, F. Fabregat-Santiago, A. Sastre-Santos and J. Bisquert, Energy Environ. Sci., 2010, 3, 1985 RSC.
- J. Bisquert and I. Mora-Seró, J. Phys. Chem. Lett., 2010, 1, 450 Search PubMed.
- E. M. Barea, C. Zafer, B. Gultekin, B. Aydin, S. Koyuncu, S. Icli, F. Fabregat Santiago and J. Bisquert, J. Phys. Chem. C, 2010, 114, 19840 CrossRef CAS.
- F. Fabregat-Santiago, J. Bisquert, G. Garcia-Belmonte, G. Boschloo and A. Hagfeldt, Sol. Energy Mater. Sol. Cells, 2005, 87, 117 CrossRef CAS.
- (a) B. C. O'Regan, I. López-Duarte, M. V. Martínez-Díaz, A. Forneli, J. Albero, A. Morandeira, E. Palomares, T. Torres and J. R. Durrant, J. Am. Chem. Soc., 2008, 130, 2906 CrossRef CAS; (b) B. C. O'Regan, K. Walley, M. Juozapavicius, A. Anderson, F. Matar, T. Ghaddar, S. M. Zakeeruddin, C. Klein and J. R. Durrant, J. Am. Chem. Soc., 2009, 131, 3541 CrossRef CAS; (c) T. Privalov, G. Boschloo, A. Hagfeldt, P. H. Svensson and L. Kloo, J. Phys. Chem. C, 2009, 113, 783 CrossRef CAS; (d) T. Marinado, K. Nonomura, J. Nissfolk, M. K. Karlsson, D. P. Hagberg, L. Sun, S. Mori and A. Hagfeldt, Langmuir, 2010, 26, 2592 CrossRef CAS.
- S. Ardo and G. J. Meyer, Chem. Soc. Rev., 2009, 38, 115 RSC.
- P. Wang, S. M. Zakeeruddin, P. Comte, R. Charvet, R. Humphry-Baker and M. Grätzel, J. Phys. Chem. B, 2003, 107, 14336 CrossRef CAS.
- E. Runge and E. K. U. Gross, Phys. Rev. Lett., 1984, 52, 997 CrossRef CAS.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B, 1988, 37, 785 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. MontgomeryJr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision A.02; Gaussian Inc.: Wallingford CT, 2009 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: additional calculation, electronic absorption, and electrochemical data. See DOI: 10.1039/c1sc00199j |
| This journal is © The Royal Society of Chemistry 2011 |