Porphyrin–hexaphyrin hybrid tapes†
Takayuki
Tanaka
a,
Naoki
Aratani
ab,
Jong Min
Lim
c,
Kil Suk
Kim
c,
Dongho
Kim
*c and
Atsuhiro
Osuka
*a
aDepartment of Chemistry, Graduate School of Science, Kyoto University, Sakyo-ku, Kyoto 606-8502, Japan. E-mail: aratani@kuchem.kyoto-u.ac.jp; osuka@kuchem.kyoto-u.ac.jp; Fax: +81-75-753-3970; Tel: +81-75-753-4008
bPRESTO, Japan Science and Technology Agency, Japan
cSpectroscopy Laboratory for Functional π-Electronic Systems and Department of Chemistry, Yonsei University, Seoul, 120-749, Korea. E-mail: dongho@yonsei.ac.kr; Fax: (+82) 2-2123-2434
First published on 19th May 2011
Abstract
A meso-meso linked porphyrin–hexaphyrin hybrid was synthesized by the cross condensation using meso-porphyrinyl-dipyrromethane and was oxidized with DDQ-Sc(OTf)3 to afford a porphyrin–[26]hexaphyrin hybrid tape that was reduced with NaBH4 to give a porphyrin–[28]hexaphyrin hybrid tape. Fully electronic conjugations in the hybrid tapes are observed in the UV/vis/NIR absorption spectra, electrochemistry, and excited-state dynamics.
In recent years, considerable attention has been focused on the exploration of π-conjugated porphyrins because of their potential applications in organic conducting materials, near-infrared dyes, molecular wires, and nonlinear optical materials.1–3 Among these, meso-meso, β-β, β-β triply-linked porphyrin arrays so-called porphyrin tapes possess a unique position in terms of extending linear planar structures, fully conjugated π-electronic networks, and exceptionally red-shifted absorption bands.4–6 Some modified variants have been also explored,7–11 including two-dimensionally extending L- and T-shaped tapes,8 C60-appended tapes,9 an amphiphilic columnar-liquid-crystal forming tape,10 and a square-shaped porphyrin sheet with a paratropic ring current.11 As a different class of important porphyrinoids, meso-aryl-substituted expanded porphyrins have also emerged, which display intriguing structural, electronic, and coordination properties.12,13Hexakis(pentafluorophenyl)-substituted [26]hexaphyrin(1.1.1.1.1.1) is the benchmark molecule in terms of its flat rectangular molecular shape and strong aromatic character.12,14 In addition, its two-electron reduced congener [28]hexaphyrin has been revealed to exist as a dynamic conformational mixture that entails twisted Möbius aromatic species as a major component.15
With these backgrounds, it occurred to us that a directly linked porphyrin–hexaphyrin hybrid is an interesting structural motif for expansion of π-conjugation. In this paper, we report the first synthesis of a meso-meso singly-linked porphyrin–hexaphyrin hybrid16 and its oxidative conversion to a meso-meso, β-β, β-β triply-linked porphyrin–hexaphyrin tape to examine how effectively the porphyrin and hexaphyrin moieties are electronically coupled and to explore the possibility to control the overall electronic property by changing the oxidation state of a hexaphyrin segment.
Results and discussion
Synthesis
meso-Porphyrinyl-dipyrromethane 2 was prepared by TFA-catalyzed condensation of meso-formylated NiII-porphyrin 1 with excess pyrrole in 72% yield.17Porphyrin–hexaphyrin hybrid 3 was synthesized by the cross condensation of 2, pentafluorobenzaldehyde, and meso-pentafluorophenyl-dipyrromethane in a ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3:2 (Scheme 1). After many experimental trials, the best yield of 3 (4.8%) was achieved by the reaction in CH2Cl2 at [2] = 33 mM12 with BF3·OEt2-catalysis and subsequent oxidation with 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ). meso-meso Linked diporphyrin 4 and triporphyrin 5 were also obtained as side products in 13.3 and 5.3% yields, respectively.18 High-resolution electrospray-ionization time-of-flight (HR-ESI-TOF) mass spectrometry revealed the parent ion peaks of 3 at m/z = 2225.6022 (calcd for C122H85F25N10Ni [M]+: m/z = 2225.6018).14 The 1H NMR spectrum of 3 in CDCl3 revealed its C2v symmetric structure, exhibiting four doublet signals both for the porphyrin protons and hexaphyrin outer βprotons (Fig. 1a, the labelling of protons were shown in Scheme 2). The inner βprotons (Hi and Hj) in the hexaphyrin segment were observed as two doublets at −2.29 and −2.42 ppm. The NH signals were not observed at room temperature probably due to broadening, but observed at −1.85 and −2.35 ppm at −20 °C. These assignments were supported by 1H–1H COSY measurement, H–D exchange experiments, and the previously reported data of meso-meso linked porphyrin arrays.5,20,21 Importantly, these data confirm that the NiII-porphyrin segment is connected at the short side of [26]hexaphyrin as a stable conformation in solution at room temperature. The UV/Vis/NIR absorption spectrum of 3, which exhibits Soret-like absorption bands at 419 and 572 nm and Q-band-like bands at 729, 892, and 1029 nm (Fig. 2), is almost identical to the sum of those of NiII-porphyrin and [26]hexaphyrin, indicating the small electronic interaction in spite of the direct connection. These data thus suggest a perpendicular conformation for 3, which has been also supported by theoretical calculations (see Figure S32 in Electronic Supplementary Information; ESI†).
:3:2 (Scheme 1). After many experimental trials, the best yield of 3 (4.8%) was achieved by the reaction in CH2Cl2 at [2] = 33 mM12 with BF3·OEt2-catalysis and subsequent oxidation with 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ). meso-meso Linked diporphyrin 4 and triporphyrin 5 were also obtained as side products in 13.3 and 5.3% yields, respectively.18 High-resolution electrospray-ionization time-of-flight (HR-ESI-TOF) mass spectrometry revealed the parent ion peaks of 3 at m/z = 2225.6022 (calcd for C122H85F25N10Ni [M]+: m/z = 2225.6018).14 The 1H NMR spectrum of 3 in CDCl3 revealed its C2v symmetric structure, exhibiting four doublet signals both for the porphyrin protons and hexaphyrin outer βprotons (Fig. 1a, the labelling of protons were shown in Scheme 2). The inner βprotons (Hi and Hj) in the hexaphyrin segment were observed as two doublets at −2.29 and −2.42 ppm. The NH signals were not observed at room temperature probably due to broadening, but observed at −1.85 and −2.35 ppm at −20 °C. These assignments were supported by 1H–1H COSY measurement, H–D exchange experiments, and the previously reported data of meso-meso linked porphyrin arrays.5,20,21 Importantly, these data confirm that the NiII-porphyrin segment is connected at the short side of [26]hexaphyrin as a stable conformation in solution at room temperature. The UV/Vis/NIR absorption spectrum of 3, which exhibits Soret-like absorption bands at 419 and 572 nm and Q-band-like bands at 729, 892, and 1029 nm (Fig. 2), is almost identical to the sum of those of NiII-porphyrin and [26]hexaphyrin, indicating the small electronic interaction in spite of the direct connection. These data thus suggest a perpendicular conformation for 3, which has been also supported by theoretical calculations (see Figure S32 in Electronic Supplementary Information; ESI†).
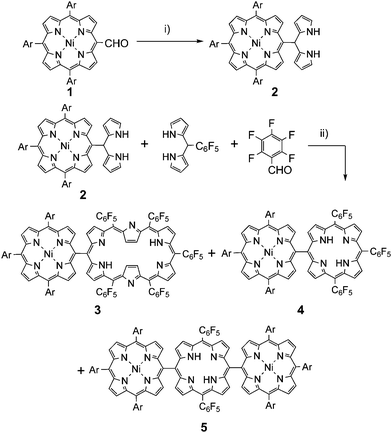 | ||
| Scheme 1 Synthesis of meso-meso singly linked porphyrin–hexaphyrin hybrid 3. Conditions: i) pyrrole, TFA; ii) BF3·OEt2, CH2Cl2, 30 min, room temperature, then DDQ, 1 h. (Ar = 3,5-di-tert-butylphenyl) | ||
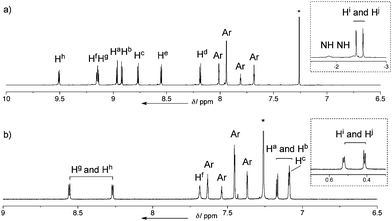 | ||
| Fig. 1 1H NMR spectra in CDCl3; a) 3 at room temperature. (Inset shows the spectrum at high field at −20 °C). b) 6 at room temperature. (*marks correspond to residual solvent peaks). | ||
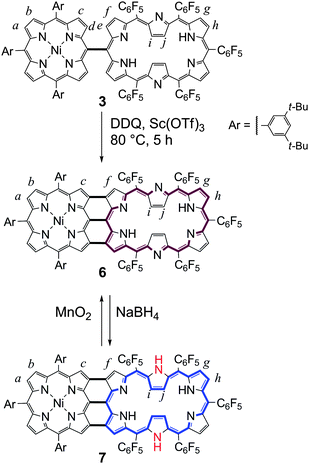 | ||
| Scheme 2 Oxidative ring-closure reaction of 3 to give 6 and redox interconversion between 6 and 7. | ||
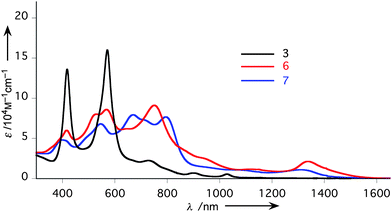 | ||
| Fig. 2 UV/Vis/NIR absorption spectra of 3 (black), 6 (red), 7 (blue) in CHCl3. | ||
Triply-linked porphyrin–[26]hexaphyrin hybrid tape 6 was obtained as dark purple solids in 72% yield in the oxidation of 3 with DDQ and Sc(OTf)3 at 80 °C for 5 h.4 The HR-ESI-TOF mass spectrum of 6 exhibited the parent-ion peak at m/z = 2219.5712 (calcd for C122H81F25N10Ni [M]−: m/z = 2219.5627). The 1H NMR spectrum of 6 in CDCl3 exhibits two doublet signals due to the hexaphyrin outer βprotons (Hg and Hh) at 8.56 and 8.27 ppm, two doublet signals due to the porphyrin outer βprotons (Ha and Hb) at 7.17 and 7.09 ppm, four signals due to the aryl protons at 7.63, 7.54, 7.45, and 7.37 ppm, and characteristically, two singlet signals due to the bay-area protons (Hc and Hf) at 7.69 and 7.08 ppm, respectively (Fig. 1b). Interestingly, two doublet signals of hexaphyrin inner βprotons (Hi and Hj) are downfield shifted to 0.52 and 0.41 ppm from the original position (−2.43 ppm) of the reference [26]hexaphyrin, suggesting a decreased diatropic ring current of 6 as compared with that of 3.
Redox interconversion
As has been studied,14,15[26]hexaphyrin can be reduced to [28]hexaphyrin with dramatic changes in the electronic and structural properties. Following this chemistry, 6 was quantitatively reduced with NaBH4 to give porphyrin–[28]hexaphyrin hybrid tape 7, which was in turn oxidized to 6 with MnO2. The HR-ESI-TOF mass spectrum of 7 exhibited the parent-ion peak at m/z = 2222.5768 (calcd for C122H83F25N10Ni [M]+: m/z = 2222.5783). In contrast to 6, the 1H NMR spectrum of 7 is solvent-polarity dependent as shown in Fig. 3. Diagnostically, the signals due to the inner βprotons (Hi and Hj), which have been assigned on the basis of the 1H–1H COSY and H–D exchange experiments, display down-field shift upon solvent change from [D6]benzene to CDCl3 and [D8]THF, suggesting increased paratropic ring currents in this order. This trend may be understood in terms of conformational constrain imposed by the direct triple linkages, which precludes a twisted Möbius aromatic conformation but favors a rectangular anti-aromatic conformation more significantly in THF due to the effective hydrogen bonding of the outer-pointing NH protons with solvent molecules.![1H NMR spectra of 7 in a) [D6]benzene, b) CDCl3, and c) [D8]THF. (*marks correspond to residual solvent peaks or solvent side peaks).](/image/article/2011/SC/c1sc00228g/c1sc00228g-f3.gif) | ||
| Fig. 3 1H NMR spectra of 7 in a) [D6]benzene, b) CDCl3, and c) [D8]THF. (*marks correspond to residual solvent peaks or solvent side peaks). | ||
In contrast to 3, porphyrin–[26]hexaphyrin hybrid tape 6 exhibits a substantially perturbed absorption spectrum with bands at 532, 566, 752, and 1333 nm and a tail reaching about 1600 nm in CHCl3, indicating the effective conjugation of the two chromophores to create a novel π-extended system (Fig. 2). Porphyrin–[28]hexaphyrin hybrid tape 7 shows a roughly similar absorption spectrum with that of 6 with bands at 399, 546, 669, 793, and 1310 nm. While the absorption spectrum of 6 is almost solvent-independent, that of 7 exhibits characteristic solvent dependence (Figure S24, 25†). The intensities of the bands around 800 and 1310 nm decrease upon increase in solvent polarity, probably reflecting the increased anti-aromatic character in the [28]hexaphyrin segment, in line with 1H NMR studies.
Electrochemistry
The electrochemical properties of hybrids 3, 6, and 7 have been studied by cyclic voltammetry (Fig. 4). The reduction potentials of 3 are similar to those of [26]hexaphyrin, indicating weak electronic communications between the two segments, but the first oxidation wave of NiII porphyrin at 0.46 V is shifted to 0.57 V probably due to the electron-withdrawing influence of a [26]hexaphyrin segment. Similarly to the porphyrin tapes,9,21 effective electronic communication is evident for hybrid tape 6 that exhibits two reversible oxidation potentials at 0.42 and 0.65 V and four reversible reduction potentials at −0.45, −0.72, −1.28, and −1.64 V. Both the first oxidation and reduction waves are shifted markedly negative and positive, respectively for about 0.15 eV. DFT molecular orbital calculation at the level of B3LYP/6-31G*(C, H, N, F)/LANL2DZ(Ni) showed nearly localized HOMO−1, HOMO, LUMO, and LUMO + 1 for 3 whose energy levels are nearly the same as those of the reference [26]hexaphyrin. On the other hand, HOMO−1, HOMO, and LUMO for 6 have been calculated to be fully delocalized over the entire hybrid system and the energy levels of HOMO and LUMO are considerably lifted and lowered, respectively, in accord with the experimental observations (Figure S34†). Reduced hybrid tape 7 shows the first and second oxidation potentials at −0.20 and 0.01 V that are distinctly lower than those of the reference [28]hexaphyrin (−0.01 and 0.15 V) and the first and second reduction waves at −0.83 and −0.94 V that are higher than those of the reference [28]hexaphyrin (−1.07 and −1.31 V). These electrochemical data are again consistent with the DFT molecular orbital calculations that predict considerably delocalized HOMO and LUMO whose energy levels are considerably lifted up from those of 6 (Figure S35†). As a consequence, the electrochemical HOMO–LUMO gaps have been determined to be 0.87 and 0.63 eV for 6 and 7, respectively, which are drastically decreased from that of 3 (1.18 eV). Noteworthy, while the electrochemical HOMO–LUMO gap of 6 is slightly smaller than the optical HOMO–LUMO gap (0.93 eV), that of 7 is considerably smaller than the optical HOMO–LUMO gap (0.95 eV) estimated from the peak positions of the Q-band-like bands. The mismatch of the HOMO–LUMO gap for 7 may be arising from the possibility that the low-energy band at 1310 nm does not correspond to HOMO–LUMO transition. This tendency has been recently revealed for anti-aromatic π-systems, in which the lowest excited states are dipole-forbidden and lying at very low energy level.22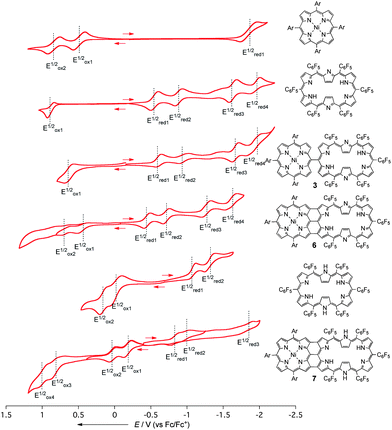 | ||
| Fig. 4 Cyclic voltammograms of 3, 6, 7, and reference compounds in CH2Cl2 with Bu4NPF6 as an electrolyte (potentials vs. Fc/Fc+). | ||
Excited state dynamics
To explore the electronic coupling between the NiII-porphyrin and hexaphyrin in 3, 6, and 7, the excited state dynamics have been investigated by femtosecond transient absorption spectroscopy. The absorption spectrum of 3 indicates only weak electronic coupling owing to the orthogonal arrangement. This feature allows us to selectively excite the constituent pigments in 3. As the excitation wavelength was tuned in resonance with the absorption bands of [26]hexaphyrin, the overall transient absorption spectra as well as their decay times of ∼120 ps largely represent the excited (π,π*) state of [26]hexaphyrin, because these features are nearly the same as those of the reference [26]hexaphyrin monomer (Figure S38†).23 On the other hand, when the excitation wavelength was largely in resonance with the Soret band of NiII-porphyrin at 400 nm, the biphasic decay was observed in the transient absorption spectra (Figure S36†). The initial fast component of ∼135 ps time constant with ∼40% in total amplitude is roughly similar to the lifetime of photoexcited [26]hexaphyrin, probably because this molecule also absorbs slightly at the excitation wavelength at 400 nm. But after the initial decay, a long-lived component with larger amplitude (∼60%) persists even at a few hundreds ps. As reported previously, the excited state dynamics of NiII-porphyrin is mainly governed by the 3(d,d) excited state with the lifetime of 250–300 ps.24 Thus, the long-lived component can be assigned as the triplet (π,π*) state of [26]hexaphyrin moiety formed through the energy transfer process from the excited 3(d,d) state of NiII-porphyrin to the 3T(π,π*) state of [26]hexaphyrin. The efficient energy transfer is probably arising from a match in the energy levels between these two states (the excited 3(d,d) and 3T(π,π*) states) and a relatively long lifetime of 3(d,d) state of NiII-porphyrin (Figure S39†).In contrast, the excited-states of 6 and 7 exhibit very fast biexponential decays with time constants of <1 ps and ∼6–7 ps (Figure S40†). These two decaying components have been assigned to the fast internal conversion process from higher excited singlet (π,π*) to the lowest 1(π,π*) state and the decay of the resultant 1(π,π*) state. Much faster excited state dynamics of 6 and 7 are probably arising from their small HOMO–LUMO gaps as a result of their fully conjugated electronic networks, similarly to the porphyrin tapes.25 Similar excited-state dynamics for 6 and 7 may indicate the dominant role of electron delocalization rather than aromatic or anti-aromatic character in the hexaphyrin segment in their excited-state decays.
Conclusions
In summary, directly linked porphyrin–hexaphyrin hybrid 3 and hybrid tapes 6 and 7 have been synthesized for the first time. In contrast to the small electronic interaction in 3, the hybrid tapes 6 and 7 show effective π-electronic delocalization, which are evident from their absorption spectra, electrochemical properties, excited-state dynamics, and molecular orbital characteristics. Due to the conformational constrain arising from the direct triple linkage, the [28]hexaphyrin segment in 7 is forced to take a planar conformation with increasing degree in polar solvents, which results in an anti-aromatic paratropic ring current.Acknowledgements
This work was supported by Grants-in-Aid (nos. 22245006 (A) and 20108001 “pi-Space”) from MEXT. T.T. acknowledges a JSPS Fellowship for Young Scientists. The work at Yonsei University was supported by World Class University Program (R-32-2008-000-1-217-0) and the Star Faculty program from the Ministry of Education and Human Resources Development, Korea, and AFSOR/AOARD grant (no. FA 2386-09-1-4092).Notes and references
- M. G. H. Vicente, L. Jaquinod and K. M. Smith, Chem. Commun., 1999, 1771 RSC; H. L. Anderson, Chem. Commun., 1999, 2323 RSC; D. P. Arnold, Synlett, 2000, 296 CAS; D. Holten, D. F. Bocian and J. S. Lindsey, Acc. Chem. Res., 2002, 35, 57 CrossRef CAS; M. O. Senge, M. Fazekas, E. G. A. Notaras, W. J. Blau, M. Zawadzka, O. B. Locos and E. M. N. Mhuircheartaigh, Adv. Mater., 2007, 19, 2737 CrossRef CAS; M. Pawlicki, H. A. Collins, R. G. Denning and H. L. Anderson, Angew. Chem., Int. Ed., 2009, 48, 3244 CrossRef CAS; N. K. S. Davis, A. L. Thompson and H. L. Anderson, Org. Lett., 2010, 12, 2124 CrossRef CAS; N. K. S. Davis, A. L. Rhompson and H. L. Anderson, J. Am. Chem. Soc., 2011, 133, 30 CrossRef CAS.
- M. J. Crossley and P. L. Burn, J. Chem. Soc., Chem. Commun., 1991, 1569 RSC; D. P. Arnold and G. A. Heath, J. Am. Chem. Soc., 1993, 115, 12197 CrossRef CAS; V. S.-Y. Lin, S. G. DiMagno and M. J. Therien, Science, 1994, 264, 1105 CrossRef CAS; L. Jaquinod, O. Siri, R. G. Khoury and K. M. Smith, Chem. Commun., 1998, 1261 RSC; M. Hoffmann, C. J. Wilson, B. Odell and H. L. Anderson, Angew. Chem., Int. Ed., 2007, 46, 3122 CrossRef.
- D. Kim and A. Osuka, Acc. Chem. Res., 2004, 37, 735 CrossRef CAS; N. Aratani, D. Kim and A. Osuka, Chem.–Asian J., 2009, 4, 1172 CrossRef CAS.
- A. Tsuda and A. Osuka, Science, 2001, 293, 79 CrossRef CAS.
- A. Tsuda, A. Nakano, H. Furuta, H. Yamochi and A. Osuka, Angew. Chem., Int. Ed., 2000, 39, 558 CrossRef CAS; A. Tsuda, H. Furuta and A. Osuka, Angew. Chem., Int. Ed., 2000, 39, 2549 CrossRef CAS; A. Tsuda, H. Furuta and A. Osuka, J. Am. Chem. Soc., 2001, 123, 10304 CrossRef CAS.
- T. K. Ahn, K. S. Kim, D. Y. Kim, S. B. Noh, N. Aratani, C. Ikeda, A. Osuka and D. Kim, J. Am. Chem. Soc., 2006, 128, 1700 CrossRef CAS; M.-C. Yoon, S. B. Noh, A. Tsuda, Y. Nakamura, A. Osuka and D. Kim, J. Am. Chem. Soc., 2007, 129, 10080 CrossRef CAS.
- I. M. Blake, A. Krivokapic, M. Katterle and H. L. Anderson, Chem. Commun., 2002, 1662 RSC; V. V. Diev, K. Hanson, J. D. Zimmerman, S. R. Forrest and M. E. Thompson, Angew. Chem., Int. Ed., 2010, 49, 5523 CrossRef CAS.
- Y. Nakamura, S. Y. Jang, T. Tanaka, N. Aratani, J. M. Lim, K. S. Kim, D. Kim and A. Osuka, Chem.–Eur. J., 2008, 14, 8279 CrossRef CAS.
- D. Bonifazi, M. Scholl, F. Y. Song, L. Echegoyen, G. Accorsi, N. Armaroli and F. Diederich, Angew. Chem., Int. Ed., 2003, 42, 4966 CrossRef CAS.
- T. Sakurai, K. Shi, H. Sato, K. Tashiro, A. Osuka, A. Saeki, S. Seki, S. Tagawa, S. Sasaki, H. Matsunaga, K. Osaka, M. Tasaka and T. Aida, J. Am. Chem. Soc., 2008, 130, 13812 CrossRef CAS.
- Y. Nakamura, N. Aratani, H. Shinokubo, A. Takagi, T. Kawai, T. Matsumoto, Z. S. Yoon, D. Y. Kim, T. K. Ahn, D. Kim, A. Muranaka, N. Kobayashi and A. Osuka, J. Am. Chem. Soc., 2006, 128, 4119 CrossRef CAS.
- J.-Y. Shin, H. Furuta, K. Yoza, S. Igarashi and A. Osuka, J. Am. Chem. Soc., 2001, 123, 7190 CrossRef CAS.
- H. Furuta, H. Maeda and A. Osuka, Chem. Commun., 2002, 1795 RSC; J. L. Sessler and D. Seidel, Angew. Chem., Int. Ed., 2003, 42, 5134 CrossRef CAS; S. Shimizu and A. Osuka, Eur. J. Inorg. Chem., 2006, 1319 CrossRef CAS.
- M. G. P. M. S. Neves, R. M. Martins, A. C. Tomé, A. J. D. Silvestre, A. M. S. Silva, V. Félix, M. G. B. Drew and J. A. S. Cavaleiro, Chem. Commun., 1999, 385 RSC.
- J. Sankar, S. Mori, S. Saito, H. Rath, M. Suzuki, Y. Inokuma, H. Shinokubo, K. S. Kim, Z. S. Yoon, J.-Y. Shin, J. M. Lim, Y. Matsuzaki, O. Matsushita, A. Muranaka, N. Kobayashi, D. Kim and A. Osuka, J. Am. Chem. Soc., 2008, 130, 13568 CrossRef CAS.
- Covalently linked porphyrin–hexaphyrin hybrids were reported: Y. Inokuma and A. Osuka, Org. Lett., 2004, 6, 3663 Search PubMed.
- K. Susumu, T. Shimidzu, K. Tanaka and H. Segawa, Tetrahedron Lett., 1996, 37, 8399 CrossRef CAS; S.-K. Hong, E. Jeoung and C.-H. Lee, J. Porphyrins Phthalocyanines, 2005, 9, 285 CrossRef CAS.
- In this reaction, a series of expanded porphyrins, especially, [26]hexaphyrin, and a trace amount of 5,15-bis(NiII porphyrin)yl-10,20,25,30-tetrakis(pentafluorophenyl)[26]hexaphyrin were obtained; see Supplementary Information†.
- For detailed characterizations of 4 and 5, see Supplementary Information†.
- A. Osuka and H. Shimidzu, Angew. Chem., Int. Ed. Engl., 1997, 36, 135 CrossRef CAS.
- L.-A. Fendt, H. Fang, M. E. Plonska-Brzezinska, S. Zhang, F. Cheng, C. Braun, L. Echegoyen and F. Diederich, Eur. J. Org. Chem., 2007, 4659 CrossRef CAS.
- M.-C. Yoon, S. Cho, M. Suzuki, A. Osuka and D. Kim, J. Am. Chem. Soc., 2009, 131, 7360 CrossRef CAS.
- T. K. Ahn, J. H. Kwon, D. Y. Kim, D. H. Jeong, S. K. Kim, M. Suzuki, S. Shimizu, A. Osuka and D. Kim, J. Am. Chem. Soc., 2005, 127, 12856 CrossRef CAS.
- D. Kim and C. Kirmaier, Chem. Phys., 1983, 75, 305 CrossRef CAS.
- H. S. Cho, D. Hong, S. Cho, D. Kim, Y. Matsuzaki, K. Tanaka, A. Tsuda and A. Osuka, J. Am. Chem. Soc., 2002, 124, 14642 CrossRef CAS; D. Bonifazi, G. Accorsi, N. Armaoli, F. Song, A. Palkar, L. Echegoyen, M. Scholl, P. Sessler, B. Jaun and F. Dieterich, Helv. Chim. Acta, 2005, 88, 1839 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details of the synthesis and spectroscopic analytical data of new compounds. See DOI: 10.1039/c1sc00228g |
| This journal is © The Royal Society of Chemistry 2011 |