Structural similarities in enzymatic, homogeneous and heterogeneous catalysts of water oxidation
Gerhard F.
Swiegers
*a,
Jack K.
Clegg
b and
Rob
Stranger
c
aIntelligent Polymer Research Institute and ARC Centre of Excellence for Electromaterials Science, University of Wollongong, Wollongong, NSW 2522, Australia. E-mail: swiegers@uow.edu.au
bDepartment of Chemistry, The University of Cambridge, Lensfield Road, Cambridge, UK CB2 1EW
cThe Research School of Chemistry, Australian National University, Canberra, ACT 0200, Australia
First published on 2nd September 2011
Abstract
A virtually identical cubical or cubane core structure appears to be a key commonality in several active homogeneous, heterogeneous and enzymatic water oxidation catalysts. We show that X-ray crystal structures of the CaMn3O4 core of the water oxidizing complex of Photosystem II coincide remarkably closely with comparable structures in a range of non-biological Co or Mn-based homogeneous and heterogeneous water oxidation catalysts. Included amongst these are molecular Co4O4 and Mn4O4 cubanes, as well as Co and Mn spinels containing cubical arrangements. This observation supports the existence of an optimum structural element for water oxidation catalysis. It also offers a potential avenue for comparing and relating homogeneous, heterogeneous and enzymatic catalysis to each other.
Introduction
Catalysis science has, for at least 100 years, sought to conceptually connect the apparently disparate fields of enzymatic, homogenous and heterogeneous catalysis.1–3 While these disciplines all involve a catalytic action, each has its own set of fundamental principles that appear to be unrelated to those of the other fields. The example of kinetics offers an illustration of the intellectual and physical distinctions that currently exist. Enzymatic kinetics has historically been formulated exclusively in terms of Michaelis–Menten behaviour, which is common in enzymes. However, such behaviour is much less widespread in non-biological molecular catalysts, so that the field of homogeneous catalysis has had to adopt a quite different formulation of kinetics. The kinetics of non-biological heterogeneous catalysts is typically unrelated to either of these; it involves expressions concerning occupancies of catalytic sites on 2-D surfaces. How, then, can catalysis in these disciplines be compared and related to one another? One way to address this question would be to find a structural element that serves as the catalytically active site for a particular reaction in any and all of homogeneous, heterogeneous and enzymatic conditions. Examination of its catalytic properties under these diverse conditions would then potentially allow one to compare catalysis in the different disciplines. Unfortunately, catalytic sites of this type are tantalizingly rare at present. Recent developments in the field of water oxidation catalysis open, however, a possible new avenue in this respect. In this work we note a remarkable similarity in the structures of several highly active enzymatic, heterogeneous and homogeneous catalysts of water oxidation. We also consider some potential implications for catalysis science.Results and discussion
Structure and structural motif of the Photosystem IIWater Oxidizing Complex
The most active and sustained water oxidation catalyst known is the Water Oxidizing Complex (WOC) of Photosystem II (PSII) that is found in photosynthetic organisms. The active site of this catalyst comprises a CaMn4O4 cluster that is preserved in essentially all species. Several single-crystal X-ray structure determinations have been carried out on the PSII-WOC. The three most detailed structures are the London structure by Barber, Iwata and colleagues (which was resolved to 3.5 Å),4 the Berlin structure by Loll, Kern and co-workers (which was resolved 2.9 Å),5 and the Osaka structure by Umena, Kawakami, Shen and Kamiya (that was resolved to 1.9 Å).6Fig. 1 depicts a previously published superimposition of the CaMn4O4 array in the London and Berlin crystal structures, looking perpendicularly down from the Ca ion to the plane of the three, core Mn ions.5 The locations of the surrounding amino acids were maintained roughly constant in this view. As can be seen, the CaMn4O4 array appears to be shifted and twisted somewhat between the two structures. The inset in Fig. 1 depicts in a side-on view, the structural arrangement and motif of the CaMn3O4 core, as it appears in ref. 4.
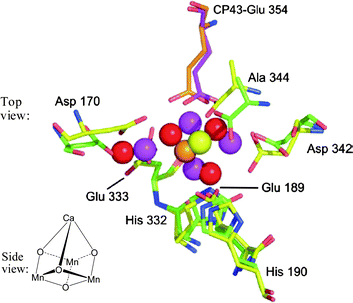 | ||
| Fig. 1 Photosystem II Water Oxidizing Complex (PSII-WOC) – best match of surrounding protein: Superimposition whilst best matching the protein framework, of the London4 and Berlin5 single-crystal X-ray structures of the core Mn4Ca cluster in the PSII-WOC. This view looks perpendicularly down from the Ca ion (yellow or orange atom) to the plane of the three core Mn ions (red or purple atoms). The yellow atom is the Ca ion and the purple atoms are the Mn ions as they appear in the London structure.4 The orange atom is the Ca ion and the red atoms are the Mn ions as they appear in the Berlin structure.5 The inset displays a side view of the CaMn3O4 core showing the cubane structural motif of the cluster according to ref. 4. (Upper image reproduced with permission from ref. 5, Copyright 2005 Macmillan Publishers Ltd, Nature Publishing Group). | ||
To examine structural variations in the CaMn3O4 core in the London, Berlin and Osaka structures, we superimposed the X-ray crystal structures of the cores in a dimensionally accurate way to achieve a best match, without regard for the location of the surrounding amino acids. Fig. 2(a) shows such a superimposition of the CaMn4O4 arrays in the London and Berlin structures. Fig. 2(b) shows a superimposition upon Fig. 2(a) of the CaMn4O4 cluster from the Osaka structure. As can be seen, the core CaMn3O4 sub-clusters are, to all intents and purposes, spatially and structurally identical in the three crystal structures. The only major difference is the location of the fourth, outlying Mn ion, which differs substantially in the London structure. The Osaka structure also contains an additional bridging O atom that links the core to the outlying, fourth Mn ion. An earlier, fourth X-ray crystal structure – the Hyogo structure – which was resolved to 3.7 Å, exhibits an identical cubane core, with the fourth Mn still further displaced.7
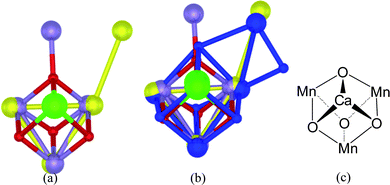 | ||
| Fig. 2 Photosystem II Water Oxidizing Complex (PSII-WOC) – Best match of the core structure: (a) superimposition of the London4 and Berlin5 single-crystal X-ray structures of the core CaMn4 cluster in the PSII-WOC without regard for the surrounding protein. This view looks perpendicularly down from the Ca ion (green atom) to the plane of the three core Mn ions (yellow or purple atoms). The purple atoms are the Mn ions as they appear in the London structure in ref. 4. The yellow atoms are the Mn ions as they appear in the Berlin structure in ref. 5. The red atoms are bridging oxygens as they appear in ref. 4. As can be seen, the core CaMn3 clusters are spatially virtually identical, with the fourth, outlying Mn ion in substantially different locations. (b) Superimposition of the recent, 1.9 Å resolution Osaka single-crystal X-ray structure6 (blue atoms) upon the structures in (a) above. As can be seen, the cubical core CaMn3 clusters in all three of the London,4Berlin5 and Osaka6 structures are spatially essentially identical. An additional bridging O atom connecting the outlying, fourth Mn ion is present in the Osaka structure. (c) The cubane structural motif of the core of the PSII-WOC. | ||
The active site of the PSII-WOC, analysed in at least four separate samples using the best available crystallographic technique, therefore clearly involves a so-called cubane, or (distorted/pyramidal) cubical core structural arrangement in which a Ca ion caps a triangle of three Mn ions. Fig. 2(c) illustrates in schematic form, the structural motif of the core cubane array. Four oxygen atoms triply bridge the Ca/Mn ions.
Given the structural consistency of the core structure as illustrated in Fig. 2(a)–(b), it is perhaps not surprising that recent computational studies have shown the London and BerlinCaMn4O4 clusters to be energetically closely related.8,9 This can reasonably be anticipated to also be the case for the Osaka structure.
The lack of evolutionary and structural diversity in this core cubane arrangement implies that combinatorial biosynthesis in Nature has yielded only this one catalytic structure capable of facilitating sustained water oxidation catalysis.
This finding begs several questions. First, why should only this one structure exist to facilitate water oxidation catalysis in Nature? Second, if this cubane structure were present in a non-biological species, would the non-biological species also catalyse water oxidation? In other words: what, precisely, is the fundamental relationship between the above cubane core structure and the facilitation of water oxidation catalysis?
In the last few years, several unrelated but apparently adventitiously parallel studies, have yielded a range of active non-biologicalwater oxidation catalysts that offer possible answers to these questions. These species, which also involve cubane active sites, include both homogeneous and heterogeneous catalysts that facilitate active water oxidation at pH 7. In this work we will note and examine these instances, and also discuss the implications that arise in respect of catalysis science.
Co-phosphate water oxidation catalyst
One of the most widely cited recent water oxidation catalysts has been the Co-phosphate system described by Nocera and Kanan.10,11 Studies have shown that this catalyst comprises of cubical arrays of the type depicted in Fig. 3(a).12 While initially believed to be a heterogeneous catalyst, it now appears that these arrays dissolve in open solution; they are therefore considered homogeneous catalysts.11c As can be seen in Fig. 3(b), the surface of the arrays involve an “open-face” checkerboard of puckered Co–O recesses and protrusions, each one of which individually forms half of an overall Co4O4cubane structure (Fig. 3(c)). Multiple layers of such arrays stacked in a lattice would necessarily contain within them, an overall cubicalCo4O4 unit cell. The structural motif of this catalyst is therefore that of a cubane, albeit a Co–O not a Mn–O one.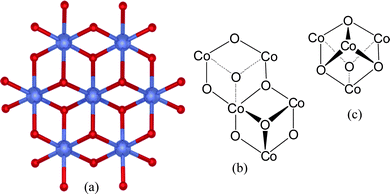 | ||
| Fig. 3 Co-phosphate (Co-phos) water oxidation catalysts: (a) structural model derived from X-ray-spectroscopy of catalytic, edge-sharing molecular cobaltate clusters according to ref. 11c. The Co ions are shown in blue. Bridging oxygen atoms are depicted in red. (b) Structural arrangement of the surface of the clusters. (c) The overall cubane structural motif of this water oxidation catalyst. | ||
How does this catalyst operate? Evidence available to date suggests that it self-assembles and dis-assembles during turnover.11 That is, Co–O cubane structures at the surface are believed to dis-assemble, releasing O2 during water oxidation.11a,b In the process, Co4+ ions appear to be converted to Co2+, which are also released to solution. Under the applied potential bias, solution Co ions re-assemble the arrays at the catalyst surface, taking up water molecules and releasing H+ and e− in proton-coupled electron transfer (PCET) reactions.11
Nanoparticulate Co3O4 spinel water oxidation catalyst
Another catalyst of interest is the AB2O4 spinel, Co3O4. In microparticulate form, this catalyst is moderately active for water oxidation.12,13 Frei and colleagues, have recently shown however, that, it becomes highly active for water oxidation in heterogeneous, nanoparticulate (nanorod) form.12The structure of the B-site of the spinel is depicted in Fig. 4(a). As can be seen, it comprises of a Co4O4cubane arrangement whose motif, shown in Fig. 4(b), is identical to that of the Co phosphate catalyst in Fig. 3(c) and the PSII-WOC in Fig. 2(b). Moreover, the surface of this heterogeneous catalyst necessarily comprises of a mixture of “open-face” A and B sites. Fig. 4(c) schematically illustrates the surface features that derive from the B site. As can be seen, they involve puckered, half-cube Co–O recesses or protrusions whose structural motif is identical to those of the Co-phosphate catalyst in Fig. 3(b).
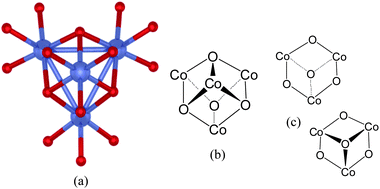 | ||
| Fig. 4 Co3O4 spinel water oxidation catalysts: (a) single-crystal X-ray structure according to ref. 13 of the B-site of Co3O4 spinel as described by Frei and co-workers.12 The Co ions are shown in blue. Bridging oxygen atoms are depicted in red. (b) The cubane structural motif of the B-site of the Co3O4 spinel. (c) Structural formations present at the surface of the Co3O4 spinel that derive from the B-site. | ||
The activity of the nanoparticulate Co3O4 spinel is substantially greater than that of the equivalent microparticulate Co3O4.12,13 Indeed, its activity is reported to exceed by 65-fold, that which could reasonably be expected, based on its larger surface area. Clearly, some factor other than surface area must impart this nanoparticulate Co3O4 with a significantly higher rate of O2 formation per surface Co than microparticulate Co3O4. Earlier studies indicated that the spinel cycles through Co4+ intermediates during water oxidation catalysis.14
Cubane model complex of the PSII-WOC
A decade ago, Dismukes and Ruettinger reported a model complex of the PSII-WOC, namely the cubane Mn4O4L6, 1 (where L = (p-MeOC6H4)2PO2− and 1 contains two Mn3+ ions and two Mn4+ ions).15Fig. 5(a) depicts the X-ray crystal structure of 1; Fig. 5(b) illustrates its structural motif.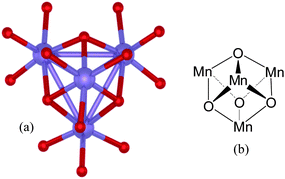 | ||
| Fig. 5 Cubane model complex: (a) single-crystal X-ray structure of the core of the model complex Mn4O4L6, 1, where L = (p-MeOC6H4)2PO2−, according to ref. 15. The ligands L bind via the free O(–P) atoms. The Mn ions are shown in blue. Bridging oxygen atoms are depicted in red. For clarity, only the ligating oxygen atoms of the phosphinate ligands are shown in the crystal structure. (b) Cubane structural motif of 1. | ||
In subsequent studies, Dismukes and co-workers showed that UV illumination (λ = 350 nm) of 1 in the gas phase resulted in the ejection of a phosphinate ligand L to form the cationic species Mn4O4L5+.15 This was followed by the spontaneous release of dioxygen, O2. Thus, 1 yielded dioxygen when illuminated with UV light in the gas phase.15
The astonishing aspect of this reaction was that the quantum efficiency of O2 release at 350 nm was found to approach 100%.15 This can only be possible if 1 facilitates an extraordinarily efficient, light-induced, collision† between bound O-reactants that has an almost 100% chance of successfully generating O–O bonds.15,16 Virtually every encounter between reactant O-atoms that is brought about by the catalyst, must lead to O–O bond formation. That is, the step of O–O bond formation, which is known to be the most critical and difficult of the steps in water oxidation catalysis, must be remarkably efficient.16
On the basis of this work and associated theoretical calculations, a mechanism of water oxidation was proposed, which is depicted in Scheme 1.15 A critical first step in this mechanism is the photo-induced loss of one of the bridging phosphinate ligands L to form the uncapped open-faced cubane structure Mn4O4L5+2. The conformational flexibility of 2 then allows the two Mn centres on the uncapped face to repel one another and, as a consequence, the two tri-μ-oxo bridging atoms on the same face are brought into reactive contact, forming, first, the peroxo (O22−) (3) and then the superoxo (O2−) species (4). Subsequent release of dioxygen results in the “open butterfly” complex Mn4O2L5+ (5).
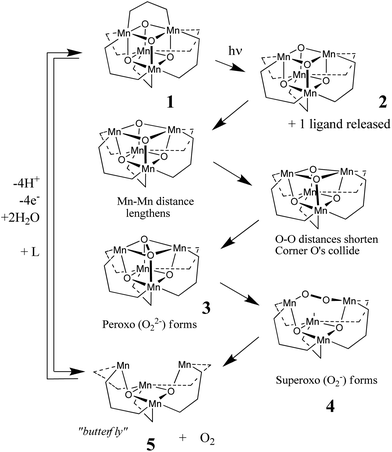 | ||
| Scheme 1 Simulation of O2 formation and release by 1 under illumination (λ = 350 nm) in the gas phase, according to Dismukes and colleagues,15 and later confirmed in DFT calculations by Musaev and co-workers.17Cluster charges have been excluded for clarity. | ||
This mechanism of O2 release has recently been subjected to a detailed computational investigation by Musaev and co-workers.17 Their DFT study at the B3LYP/Lanl2dz level of theory confirmed that the key step of O–O bond formation from the uncapped cubane structure (2) to form the peroxo species (3), proceeds via a rate-determining barrier of around 120 kJ mol−1. This barrier appears to derive essentially only from the strain of conformation flexing, not from the thermodynamics of the O–O collision.† During this process, the two Mn centres on the lower face of the cubane are reduced from the 4+ to the 3+ oxidation state. Following this, the cubane structure undergoes significant flattening to form, first, a superoxo intermediate in which O2− is bound between two Mn centres – one from the upper face of the former cubane and the other from the lower face – and then a second intermediate in which neutral O2 is bound to only one Mn centre, originally from the upper face of the cubane. The calculated barriers accompanying these intermediate steps were found to be insignificant. Subsequent release of dioxygen results in formation of the Mn4O2L5+ complex 5 in which two of the Mn centres are in the 3+ oxidation state and the other two are in the 2+ state. The overall reaction from the uncapped cubane (2) to dioxygen elimination and the “open butterfly” complex (5) was calculated to be exothermic by ca. 60 kJ mol−1.
Separate studies revealed that in solution, 1 is capable of reversibly abstracting 4H+ + 4e−via PCET processes and releasing 2 H2O molecules, to form a “pinned butterfly” species Mn4O2L6 that is, effectively 5, with one extra bridging ligand L.15 That is, a pathway potentially exists from 5 back to 1. This pathway (shown on the left of Scheme 1) involves 5 taking up a sixth ligand L and 2 H2O to yield 1, with accompanying release of 4 H+ and 4 e− by PCET reactions. In other words, a possible overall photocatalytic cycle exists in which 1 photo-generates O2 and is then recycled by taking up 2 H2O and releasing 4 H+ + 4 e−.
Water oxidation catalysis involving 1
In later work, Spiccia, Dismukes, Swiegers and co-workers ion-exchanged cubane1++ into a thin layer of Nafion deposited on a conducting surface.18 When biased at 1.20 V (vs.SHE) and illuminated with sunlight, the 1++/Nafion layer rapidly and readily catalysed the conversion of H2O into O2, with accompanying H2 formation at the other electrode.18 The catalytic effect, which was substantial and sustained, occurred at a voltage similar to that employed by the PSII-WOC, which is 1.25 V (vs.SHE).The mechanism was proposed to be comparable to that in Scheme 1. That is, it was suggested to involve a light-induced release of a single phosphinate ligand to form an open face cube (2) in the Nafion, followed by a dis-assembly and re-assembly process in which the Mn4O4cubane was broken up by O2 release (to 5) and then re-assembled by incorporation of two, new H2O molecules with the loss of 4H+ and 4e− in PCET steps (to 1).19 As 1 undergoes dynamic self-assembly, MnII may be released.
While 1++/Nafion has no light-harvesting system and can therefore not be nearly as vigorous a catalyst as the PSII-WOC, it was found nonetheless, to be one of the most active and durable man-made, homogeneous water oxidation photocatalysts.19 The Nafion support appeared to be critical insofar as it may:19,20 (a) facilitate the transport of H+ products away from the reaction, and (b) trap 1++ in its hydrophobic pockets during the proposed assembly-disassembly process during turnover.
In subsequent work, 1++/Nafion was adapted to operate in a free-standing, water-splitting dye-sensitized solar cell. The cell was free-standing because it did not need an externally applied potential to operate.21 The photoanode of the cell was prepared by attaching a Ru(II) dye, [RuII(bipy)2(bipy(COO)2)] (bipy = 2,2′-bipyridyl), onto a titania-coated FTO conductive glass electrode, followed by overcoating with a thin Nafion membrane into which 1++ was introduced by ion-exchange.21 Upon illumination with visible light while immersed in water and connected to a Pt counter electrode, the resulting photoanode spontaneously oxidized H2O to release O2 gas and protons to solution. An electrical current simultaneously flowed through the external circuit, to reduce protons in the solution to H2 at the other electrode. Thus, the cell spontaneously split water into O2 and H2 upon illumination with light; no other energy source was needed.
Despite being far from optimized, the cell generated a potential of 0.6 V in air, was active under illumination of up to λ < 625 nm, and achieved an IPCE of 1.7% at λ = 450 nm.21 There was no need for an externally applied voltage to drive the system because the oxidized RuIII form of the photoanode dye that is created by absorption of a photon, and the reduced form of the cubane catalyst 5 that is created by O2 release, mutually cycle each other during turnover.21 This photoanode arguably represents a functional analogue of the PSII-WOC.17,20,21 Like the earlier-mentioned Co-phosphate catalyst, the 1++/Nafion system is capable of splitting non-potable water, like seawater, into pure H2 and O2, with no observable Cl2 formation.22
A report that appeared too late to be considered in this study describes a molecular Co4O4 cubane analogue to 1 that is more active in water oxidation catalysis.23
Birnessite water oxidation catalyst
Spiccia and colleagues have recently reported that nanoparticles of the Mn–O mineralbirnessite may be formed in the 1++/Nafion under suitable conditions.24 Moreover, the birnessite nanoparticles appear to be capable of actively catalyzing the oxidation of H2O into O2.Birnessite has a layered structure that comprises stacked, 2-D strata of hetero-ions (which may be Ca, Mg, K, and the like) interleaved with 2-D extended Mn–O sheets.25 Birnessite has previously been hypothesized to be the evolutionary origin of the PSII-WOC.26
Fig. 6(a) depicts an X-ray structure of an extended Mn–O sheet in birnessite. Fig. 6(b) shows the atom connectivity of these sheets. As can be seen, they comprise puckered, half-cube Mn–O recesses and protrusions that are distinctly similar to those depicted in Fig. 3(a) and Fig. 4(c). The structural motif of birnessite is, consequently, also a cubane, being identical to that of the PSII-WOC (Fig. 1 and 2), the Co-phosphate Co4O4catalyst (Fig. 3), the B-site of nanoparticulate Co3O4 spinels (Fig. 4) and the Mn4O4cubane model complex 1 (Fig. 5).
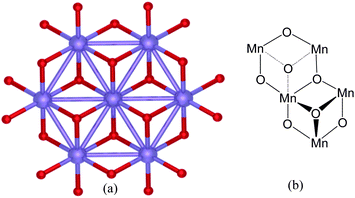 | ||
| Fig. 6 Birnessite water oxidation catalysts: (a) single-crystal X-ray structure according to ref. 25 of a Mn–O extended sheet layer in K birnessite. The Mn ions are shown in blue; bridging oxygen atoms are depicted in red. Birnessite structures have been reported to be catalytically active for water oxidation.24 (b) The structural arrangement of the Mn–O sheet layers in birnessite. (Hetero-ions in layers above and below the Mn–O layer, have been omitted for clarity). | ||
λ-Mn2O4 spinel water oxidation catalyst
In recent work, Dismukes and co-workers have shown that, while the AB2O4-type spinel LiMn2O4, is inactive in the facilitation of water oxidation, it becomes a highly active catalyst when the Li+ ion is removed from the A-site.27 The remaining λ-Mn2O4 contains a Mn4O4cubane structure in its B-site.27Fig. 7(a) depicts the crystal structure of the B-site of λ-Mn2O4 spinel.28Fig. 7(b) shows the atom connectivity of the B-site within the bulk spinel lattice. As can be seen, the B-site displays a cubane structural motif. Fig. 7(c) shows the features that are present at the surface of the spinel due to the B site. These are puckered Mn–O half-cubes; their similarity to surface features in previous figures is unmistakable.
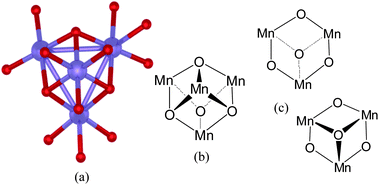 | ||
| Fig. 7 λ-MnsO4 spinel water oxidation catalysts: (a) single-crystal X-ray structure according to ref. 28 of the B-site of λ-MnsO4 spinel as described by Dismukes and co-workers.27Mn ions are shown in blue; bridging oxygen atoms are depicted in red. (b) The cubane structural motif of the B-site of the Li-free λ-MnsO4 spinel. (c) Structural formations at the surface of the λ-MnsO4 spinel that derive from the B-site. | ||
The key property of this spinel is that it displays no catalytic activity whatsoever when Li ions are present in the A-site. However, when the Li ions are removed, freeing up the shared μ3-O atoms in the B-site for motion, it becomes a highly active catalyst.27 Thus, it appears that introducing enhanced motion in the μ3-O atoms of the surface B-sites, may create the catalysis.27 This effect seems similar to that created in 1 by the loss of a single capping ligand to yield a flexible open face during turnover.19 That is, it may involve a dis-assembly and re-assembly process in which the Mn4O4cubane structure at the surface of the catalyst is broken up by μ3-O collision across the face, yielding O2, and then re-assembled by incorporation of two, new H2O molecules in PCET processes (with accompanying loss of 4 H+ and 4 e−).
Such a scenario potentially also explains why the nanoparticulate Co3O4 spinel of Frei and co-workers13 is so disproportionately more active than its microparticulate counterpart. The extent of motion by atoms in a nanoparticle lattice can reasonably be expected to be greater than in a microparticle lattice. This is because the lattice contains fewer atoms in total and must therefore be less rigid. Surface defects, which are also more prevalent in nanoparticles, would, arguably, amplify this effect. Enhanced oscillatory, thermal motion by the bridging μ3-oxo atoms within the constraints of the crystal lattice may therefore potentially explain the anomalously high catalytic rate per surface Co ion in the Co3O4 nanoparticles relative to the equivalent microparticles.
It has long been recognized that defects and vacancies play a critical role in the surface activity of heterogeneous catalysts. One reason for this, at least in these solid-state water oxidation catalysts, is now implied to be because they allow for greater motion in the surface atoms of the catalyst.
Polyoxotungstate water oxidation catalyst
A final, potentially notable homogeneous water oxidation catalyst is the complex of Hill and co-workers', [Co4(H2O)2(PW9O34)]10−6.29 This species comprises of the core depicted in Fig. 8(a) stabilized by oxidatively resistant bulky polyoxotungstate ions. The complex undergoes dynamic self-assembly in water to yield the highest recorded turnover frequency of any abiological catalyst for catalyticwater oxidation (>5 s−1 at pH 8).29 While the mechanism of catalysis by 6 is unknown, its core structural motif in the solid state is, effectively, a cubane, being identical to the cubical structures in Fig. 3(b), 4(c), 5(b), 6(b) and 7(b).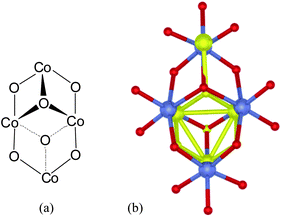 | ||
| Fig. 8 Polyoxotungstate water oxidation catalyst: (a) the structural arrangement of the core of the polyoxotungstate water oxidation catalyst 6.29Co ions are shown in blue; bridging oxygen atoms are depicted in red. (b) Single-crystal X-ray structure of the core of 6 (blue atoms = Co, red atoms = O)29 overlaid with the single-crystal X-ray structure of the CaMn3O4 core of the of the London structure of the PSII-WOC according to ref. 4 (yellow structure). The Ca ion in the PSII-WOC has been excluded for clarity. As can be seen, the core structures overlap to all intents and purposes. The outlying, fourth Mn ion in the PSII-WOC coincidentally overlaps the fourth Co ion in 6. | ||
The self-assembling character of 6 is consistent with the presence of a library of possible component structures in solution. This could theoretically include open-faced arrangements.
The physical structure of the core of 6 in the solid state is remarkably similar to the structure of the PSII-WOC. To show just how closely related these species are, Fig. 8(b) superimposes in a dimensionally accurate way, the X-ray structure of the core of 6 upon the London structure of the CaMn3O4 core of the PSII-WOC from Fig. 2(a). As can be seen, the core structures are virtually co-incident. The outlying, fourth Mn ion in the London structure of the PSII-WOC inadvertently also happens to overlap with the fourth Co in the core of 6; this is not true for the Berlin and Osaka structures.
An apparent convergence in the structures and structural motifs of water oxidation catalysts
The species discussed above represent some (albeit not all)30 of the current “state-of-the-art”, high-performance, abiological homogeneous and heterogeneous catalysts of water oxidation at pH 7. They also display an astonishing and noteworthy structural coincidence with the CaMn3O4 core of the PSII-WOC as revealed by X-ray crystallography. Fig. 9 and 10 illustrates the extent of this similarity.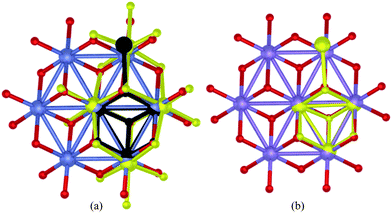 | ||
| Fig. 9 Superimposition of: (a) three water oxidation catalysts: (i) the single-crystal X-ray structure of the core of 6 (yellow structure),29 (ii) the single-crystal X-ray structure of the CaMn3O4 core of the London structure of the PSII-WOC according to ref. 4 (black structure) (The Ca ion in the PSII-WOC has been excluded for clarity) and: (iii) the X-ray-spectroscopy derived structural model of the edge-sharing molecular cobaltate clusters according to ref. 11c where Co ions are shown in blue and bridging oxygen atoms are depicted in red. As can be seen, the core structures coincide to all intents and purposes. The outlying, fourth Mn ion in the PSII-WOC coincidentally overlaps the fourth Co ion in 6. (b) two water oxidation catalysts: (i) the single-crystal X-ray structure of the CaMn3O4 core of the London structure of the PSII-WOC according to ref. 4 (yellow structure) (The Ca ion in the PSII-WOC has been excluded for clarity) and: (ii) the single-crystal X-ray structure according to ref. 25 of the surface of a Mn–O layer in K birnessite, where Mn ions are shown in blue and bridging oxygen atoms are depicted in red. The outlying, fourth Mn ion in the PSII-WOC coincidentally overlaps a neighbouring Mn ion in the birnessite. | ||
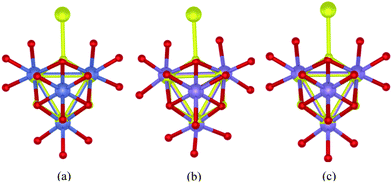 | ||
| Fig. 10 Superimposition of: (a) (i) the single-crystal X-ray structure of the CaMn3O4 core of the London structure of the PSII-WOC according to ref. 4 (yellow structure) (The Ca ion in the PSII-WOC has been excluded for clarity) and: (ii) single-crystal X-ray structure according to ref. 13 of the B-site of Co3O4 spinel as described in ref. 12, where Co ions are shown in blue and bridging oxygen atoms are red. (b) (i) the single-crystal X-ray structure of the CaMn3O4 core of the London structure of the PSII-WOC according to ref. 4 (yellow structure) (The Ca ion in the PSII-WOC has been excluded for clarity) and: (ii) the single-crystal X-ray structure according to ref. 15 of the model complex Mn4O4L6, 1, where L = (p-MeO-C6H4)2PO2−. Mn ions are shown in blue; bridging oxygen atoms are red. As can be seen, the core structures coincide to all intents and purposes. (c) (i) the single-crystal X-ray structure of the CaMn3O4 core of the London structure of the PSII-WOC according to ref. 4 (yellow structure) (The Ca ion in the PSII-WOC has been excluded for clarity) and: (ii) the single-crystal X-ray structure according to ref. 28 of the B-site of λ-MnsO4 spinel as described in ref. 27, where Mn ions are shown in blue and bridging oxygens atoms are in red. As can be seen, the core structures coincide to all intents and purposes. | ||
Fig. 9(a) superimposes in a dimensionally accurate way, the X-ray structures of the cores of 6 and the London PSII-WOC in Fig. 8(b), upon the structure of the Co-phosphate catalyst in Fig. 3(a).
In Fig. 9(b), the CaMn3O4 core of the London structure of the PSII-WOC is superimposed upon the sheet structure of birnessite illustrated in Fig. 6.
In Fig. 10, the CaMn3O4 core of the London structure of the PSII-WOC is accurately superimposed upon:
(i) Fig. 10(a): the B-site of Co3O4 spinel from Fig. 4(a),
(ii) Fig. 10(b): the cubane model complex 1 from Fig. 5(a), and
(iii) Fig. 10(c): the B-site of λ-MnsO4 spinel from Fig. 7(a).
As can be seen in Fig. 9 and 10, in all cases there is an astonishingly close match of the core of the non-biological homogeneous and heterogeneous catalysts with the Mn3O3 portion of the core of the PSII-WOC. In most cases, they are effectively indistinguishable.
Table 1 lists a statistical goodness-of-fit comparison of the positional overlays shown in Fig. 8–10. The comparison comprises a root mean square (RMS) measurement of consistency in three metals and their bridging O. As can be seen in Table 1, there is statistically very little variation in the depicted structures. Indeed, the high-resolution Osaka structure of the PSII-WOC is statistically more closely coincident with most of the non-biological catalysts than it is with the London structure of the PSII-WOC. In other words, there is more variation within the X-ray structures of the PSII-WOC than there is with most of the non-biological catalysts.
| Fig. | 2(a) | 2(a) | 2(c) | 3 | 4 | 5 | 6 | 7 | 8 |
|---|---|---|---|---|---|---|---|---|---|
| London | Berlin | Osaka | Co-phos | Co3O4 | 1 | Birnessite | λ-Mn2O4 | 6 | |
| London | — | 0.17 | 0.255 | 0.0632 | 0.0893 | 0.121 | 0.0995 | 0.0797 | 0.296 |
| Berlin | 0.17 | — | 0.0757 | 0.167 | 0.158 | 0.162 | 0.153 | 0.158 | 0.251 |
| Osaka | 0.255 | 0.0757 | — | 0.262 | 0.235 | 0.219 | 0.245 | 0.237 | 0.272 |
| Co-phos | 0.0632 | 0.167 | 0.262 | — | 0.0363 | 0.100 | 0.0747 | 0.0692 | 0.239 |
| Co3O4 | 0.0893 | 0.158 | 0.235 | 0.0363 | — | 0.0417 | 0.0209 | 0.0109 | 0.214 |
| 1 | 0.121 | 0.162 | 0.219 | 0.100 | 0.0417 | — | 0.0374 | 0.0506 | 0.189 |
| Birnessite | 0.0995 | 0.153 | 0.245 | 0.0747 | 0.0209 | 0.0374 | — | 0.0227 | 0.203 |
| λ-Mn2O4 | 0.0797 | 0.158 | 0.237 | 0.0692 | 0.0109 | 0.0506 | 0.0227 | — | 0.225 |
| 6 | 0.296 | 0.251 | 0.272 | 0.239 | 0.214 | 0.189 | 0.203 | 0.225 | — |
Table 2 lists several pertinent bond angles and lengths in the respective structures in Fig. 8–10. The extent of similarity is, again, notable. For example, with few exceptions, the M–O bond lengths about the central μ3oxygen in the above species fall in the range 1.82–1.94 Å; the O–M–O bond angles lie about 82–87°.
| M–M (Å) | M–O (Å) | M–O–M (°) | O–M–O (°) |
|---|---|---|---|
| London (Fig. 2(a)) | |||
| 2.657, 2.677, 2.722 | 1.865, 1.774, 1.948, 1.887, 1.809, 1.853, 1.821, 1.823, 1.830 | 93.77, 90.43, 93.95, 93.45, 93.41, 96.35 | 93.05, 91.85, 91.57, 87.61, 87.36, 85.12, 85.10, 87.51, 85.61 |
| Berlin (Fig. 2(a)) | |||
| 3.131, 2.787, 2.691 | N/A | N/A | N/A |
| Osaka (Fig. 2(c)) | |||
| 3.298, 2.912, 2.760 | 1.937, 2.064, 2.030, 1.791, 2.605, 2.398, 1.832, 2.126, 2.019 | 93.36, 92.28, 82.38, 89.21, 88.09, 117.76 | 93.54, 92.92, 87.81, 84.66, 80.32, 95.46, 76.57, 78.69, 90.99 |
| Co-phosphate (Fig. 3) | |||
| 2.795, 2.798, 2.813 | 1.912, 2.050, 1.888, 1.887, 2.024, 1.911, 1.898, 1.937, 1.896 | 89.78, 90.46, 96.34, 95.71, 93.57, 93.78 | 93.48, 92.20, 95.11, 83.98, 83.96, 86.39, 89.06, 89.64, 86.79 |
| Co3O4 (Fig. 4) | |||
| 2.858, 2.858, 2.858 | 1.923, 1.923, 1.923, 1.923, 1.923, 1.923, 1.923, 1.923, 1.923 | 95.95, 95.95, 95.95, 95.95, 95.95, 95.95 | 83.72, 83.72, 83.72, 83.72, 83.72, 83.72, 83.72, 83.72, 83.72 |
| 1 (Fig. 5) | |||
| 2.926, 2.926, 2.926 | 1.997, 1.909, 1.997, 1.909, 1.997, 1.909, 1.929, 1.929, 1.929 | 96.99, 96.99, 96.99, 96.99, 96.99, 96.99, 98.63, 98.63, 98.63 | 83.01, 83.01, 83.01, 82.77, 80.51, 82.77, 80.51, 82.77, 80.51 |
| Birnessite (Fig. 6) | |||
| 2.846, 2.898, 2.898 | 1.926, 1.926, 1.951, 1.926, 1.926, 1.951, 1.926, 1.926, 1.952 | 96.74, 96.74, 95.25, 96.74, 96.74, 95.26 | 96.74, 96.74, 95.25, 83.26, 83.26, 84.74, 83.26, 83.26, 84.74 |
| λ-Mn2O4 (Fig. 7) | |||
| 2.839, 2.839, 2.839 | 1.907, 1.907, 1.907, 1.907, 1.907, 1.907, 1.907, 1.907, 1.907 | 96.18, 96.18, 96.18, 96.18, 96.18, 96.18 | 83.46, 83.46, 83.46, 83.46, 83.46, 83.46, 83.46, 83.46, 83.46 |
| 6 (Fig. 8) | |||
| 3.212, 3.294, 3.209 | 2.258, 2.215, 2.085, 2.084, 2.124, 2.087, 2.215, 2.258, 2.199 | 94.84, 99.42, 100.69, 93.29, 92.19, 94.84 | 83.22, 85.93, 87.71, 82.88, 84.25, 82.48, 85.16, 85.16, 83.63 |
Thus, there appears to be something of a convergence in both the structural motifs as well as major elements of the actual physical structure in these homogeneous, heterogeneous and enzymatic catalysts of water oxidation. All involve M′M3O4-cubane type cores (M′ = Co, Mn, Ca; M = Co, Mn). The M3O3 portions of the cores appear to be essentially structurally coincident in all of the examples discussed above.
This commonality may also extend to their catalytic actions. Thus, several of these catalysts appear to undergo spontaneous, dis-assembly and re-assembly of the cubane, at an open face (or surface) during catalysis. Co4+/Mn4+ ions may be involved in the catalytic formation of O2, with the resulting Co2+/Mn2+ ions taking up water molecules to dynamically re-assemble the catalyst with accompanying release of H+ + e− in PCET steps.
The PSII-WOC also harnesses PCET steps and has been proposed to utilize an open face that is formed during conformational flexing of the enzyme.8,9,32Water molecules are thought to bind, bridge and exchange at the open face, after which continuing conformational change is proposed to bring them into reactive contact with each other.
These apparent commonalities in an otherwise disparate and unconnected range of homogeneous, heterogeneous and enzymatic catalysts are remarkable. Studies appear to be confirming the findings of combinatorial bio-synthesis regarding the utility of the cubane structure in water oxidation catalysis.
Implications of the commonality – an optimum approach trajectory?
These results answer one of the two questions that were raised at the beginning of this article, namely the question as to whether a non-biological species which contains a cubane structure similar to that of the PSII-WOC, would display water oxidation catalysis? We can now definitively answer “yes” to that question for both homogeneous and heterogeneous forms of abiological catalysis, with the apparent provisos that:(1) the cubane structure involves four Co or four Mn ions bridged by four μ3-O atoms, or it comprises an open-faced cubical subset thereof,
(2) an open face exists in the cubane structure during at least one point of the catalytic cycle. It may further be necessary that this open face be framed, either directly or indirectly, by two Co or Mn ions in the 4+ state, and
(3) the relevant μ3-O atoms may require sufficient freedom of motion within the constraints of the cubical molecular or lattice structure. This motion may be either conformational motion in molecular, homogeneous systems, or oscillatory thermal motion in extended, heterogeneous systems.
While this answers the above question, it also raises a new question: why should a cubane structure on the surface of a heterogeneous catalyst display activity that is qualitatively similar to a cubane structure in the molecular framework of a homogeneous catalyst and to a cubane structure in the proteinaceous framework of an enzymatic one?
This query is relevant to the second of the two questions posed at the beginning of this article, namely: why does only one structure – a cubane structure – facilitate water oxidation catalysis in biology? We can expand that question to ask: why does a cubane structure facilitate water oxidation catalysis in biological enzymatic species as well as non-biological homogeneous and heterogeneous species?
We cannot definitively answer this important question at present. However, it is clear that all of these catalysts must share a common fundamental attribute that favours catalyticwater oxidation.33 One possible attribute is that all of these species employ a common approach trajectory in the way that their bound oxo-reactants combine to form O2.33
For any transformation involving two reactants coming into contact and reacting with each other, there will generally be only one optimum approach pathway. That is, there will be one ideal trajectory in space along which the reactant atoms must approach each other and come into contact, in order to have the greatest likelihood of undergoing reaction. This optimum trajectory will necessarily be the same regardless of whether the catalysis occurs in an enzymatic, homogeneous or heterogeneous setting.33
In the case of water oxidation yielding O2, we have a possible indication of such an optimum pathway from the earlier-mentioned O2 evolution studies on 1 in the gas phase under UV light.15,17 In that work it was found that two opposing, bridging μ3-O atoms approaching each other and coming into contact across an open face of the cubane (framed by Mn3+/4+ ions), yielded O2 with essentially 100% certainty (according to the quantum efficiency measured for 1 in the gas phase under 350 nm illumination).15,16
This mechanism, which on the basis of calculations is viable for 1,17 is also attractive for most of the other catalytic systems discussed here. Thus, all of the extended cubane structures possess open faces (or surfaces) in which the tri-μ-oxo bridges between the Mn or Co centres appear to have sufficient freedom of motion to approach and come into contact with one another.
This potentially predisposes these systems to O–O bond formation in a manner similar to that described above for 1. Moreover, it concurs with the studies on the λ-Mn2O4 spinel and Co3O4 spinel described above, which suggest that freedom of motion in the μ3-oxo bridges of those species may be critical to the catalytic effect.15,27 In other words, whereas two tri-μ-oxo species are brought into reactive encounter across the open face of 1 by the conformational flexing of the cubane structure (Scheme 1), they may, in the solid-state catalysts, be brought into collision across the open face (or surface) by oscillatory thermal motion. Such a mechanism would, furthermore, be consistent with earlier studies showing that the Co ions in the Co-phosphate catalyst and Co3O4 spinels cycle through 4+ intermediates during water oxidation catalysis.11,14 This analogy could potentially also extend to the PSII-WOC in that computational studies8,9,32 have indicated that the substrate waters may be accommodated in the cleft region formed between the Ca and the two Mn centres at the open face of the CaMn3O4 cubane structure.
Thus, it seems possible that the fundamental relationship between the cubane structure and water oxidation catalysis may lie in the fact that the cubane structure constrains its bridging μ3-O atoms to approach each other along a pathway which has an apparent near-100% likelihood of reaction.33
This would explain why the cubane structure leads to catalytic activity in heterogeneous and homogeneous, as well as enzymatic forms. It also offers a plausible explanation for why combinatorial biosynthesis has led to a cubane active site in the PSII-WOC: the cubane may be the most efficient structure for creating O2 from reactant O atoms that derive from water.
Disclaimer: further studies are needed
In conclusion, we should note that any inferences regarding the structural similarities described above must be considered tentative at this stage. There are still too many uncertainties regarding, for example, the possibility of radiation-induced reduction of the Mn ions in the X-ray structures of the PSII-WOC. Lower oxidation state clusters may, conceivably, better represent the actual enzyme data. As noted in Table 2, minor errors were also found in the original data for some of the X-ray crystal structures.Similarly, water oxidation is known to occur by several different catalytic mechanisms,34 of which the above proposal is only one. A range of catalysts with radically different structures to a cubane are also active in water oxidation catalysis.30 The fact that a cubane structure may facilitate a particularly efficient catalytic reaction step does not mean that such species will necessarily generate the highest overall activity. Overall catalytic activity may be influenced by numerous other factors, including, for example, binding affinity for the reactants and products, as well as the vigour of conformational or oscillatory thermal motion.
A proper consideration of the catalytic mechanisms of the above catalysts requires a detailed comparison of their electronic as well as their structural similarities. Such a study is beyond the scope of the present work. Previous workers have examined this issue in only an incidental way. Thus, Sun and co-workers have performed plane-wave DFT studies35 on the release of lattice oxygen from birnessite, which is of relevance for catalytic cycles involving the release or incorporation of oxygen. The thermochemistry indicated that radical oxygen release is the preferred mode of oxygen vacancy formation. However, oxide release becomes somewhat less energetically unfavourable in highly negatively charged lattices. Plane-wave DFT studies examining the band gaps and band structures of the MnO2 sheets in the birnessite structure have also been reported by Kwon et al.36 Musaev et al. have performed DFT studies37 on the tetra-ruthenium analogue (as well as four oxidized variants thereof) of the polyoxotungstate complex 6. Solvent corrections were employed to compensate for the highly anionic character of the species investigated (total charges ranging from −6 to −10) that are normally quenched in the solid state by rubidium and potassium counterions. Sartorel et al. have also performed computational studies38 on the ruthenium polyoxotungstate cluster and drawn a strong connection between this core complex and the water oxidising complex of PSII, exploring an analogous five-state redox cycle in the catalyst.
Acknowledgements
JKC acknowledges the support of the Marie Curie IIF scheme of the 7th EU Framework Program.Notes and references
- J. Bercaw in Opportunities for Catalysis Science in the 21st Century. A Report from the Basic Energy Sciences Advisory Committee (Workshop Chair: J. M. White), May 14–16, 2002, US Department of Energy Search PubMed.
- J. Meurig Thomas and R. J. P. Williams, Philos. Trans. R. Soc. London, Ser. A, 2005, 363, 765–791 CrossRef.
- A. Corma, Catal. Rev., 2004, 46, 369–417 CAS.
- K. N. Ferreira, T. M. Iverson, K. Maghlaoui, J. Barber and S. Iwata, Science, 2004, 303, 1831–1838 CrossRef CAS.
- (a) B. Loll, J. Kern, W. Saenger, A. Zouni and J. Biesiadka, Nature, 2005, 438, 1040–1044 CrossRef CAS; (b) J. Yano, J. Kern, K.-D. Irrgang, M. J. Latimer, U. Bergmann, P. Glatzel, Y. Pushkar, J. Biesiadka, B. Loll, K. Sauer, J. Messinger, A. Zouni and V. K. Yachandra, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 12047–12052 CrossRef CAS.
- Y. Umena, K. Kawakami, J.-R. Shen and N. Kamiya, Nature, 2011, 473, 55–60 CrossRef CAS.
- N. Kamiya and J.-R. Shen, Proc. Natl. Acad. Sci. U. S. A., 2002, 100, 98–103 CrossRef.
- S. Petrie, R. Stranger and P. J. Pace, Chem.–Eur. J., 2008, 14, 5482–5494 CrossRef CAS.
- S. Petrie, R. Stranger, P. Gatt and R. J. Pace, Chem.–Eur. J., 2007, 13, 5082–5089 CrossRef CAS , and references therein.
- M. W. Kanan and D. G. Nocera, Science, 2008, 321, 1072–1075 CrossRef CAS.
- (a) D. A. Lutterman, Y. Surendranath and D. G. Nocera, J. Am. Chem. Soc., 2009, 131, 3838–3839 CrossRef CAS; (b) G. McAlpin, Y. Surendranath, M. Dinca, T. A. Stich, S. A. Stoian, W. H. Casey, D. G. Nocera and R. D. Britt, J. Am. Chem. Soc., 2010, 132, 6882–6883 CrossRef; (c) M. W. Kanan, J. Yano, Y. Surendranath, M. Dinca, V. Yachandra and D. G. Nocera, J. Am. Chem. Soc., 2010, 132, 13692–13701 CrossRef CAS , and references therein.
- F. Jiao and H. Frei, Angew. Chem., Int. Ed., 2009, 48, 1841–1844 CrossRef CAS , and references therein.
- X. Liu and C. T. Prewitt, Phys. Chem. Miner., 1990, 17, 168–172 CrossRef CAS.
- R. N. Singh, J.-F. Koenig, G. Poillerat and P. Chartier, J. Electrochem. Soc., 1990, 137, 1408–1413 CrossRef CAS.
- (a) W. Ruettinger, M. Yagi, K. Wolf, S. Bernasek and G. C. Dismukes, J. Am. Chem. Soc., 2000, 122, 10353–10357 CrossRef CAS; (b) M. Yagi, K. V. Wolf, P. J. Baesjou, S. L. Bernasek and G. C. Dismukes, Angew. Chem., Int. Ed., 2001, 40, 2925 CrossRef CAS; (c) W. Ruettinger, C. Campana and G. C. Dismukes, J. Am. Chem. Soc., 1997, 119, 6670–6671 CrossRef CAS.
- G. F. Swiegers, J. Huang, R. Brimblecombe, J. Chen, G. C. Dismukes, U. T. Mueller-Westerhoff, L. Spiccia and G. G. Wallace, Chem.–Eur. J., 2009, 15, 4746–4759 CrossRef CAS.
- A. E. Kuznetsov, Y. V. Geletii, C. L. Hill and D. G. Musaev, J. Phys. Chem. A, 2010, 114, 11417–11424 CrossRef CAS.
- R. Brimblecombe, G. F. Swiegers, G. C. Dismukes and L. Spiccia, Angew. Chem., 2008, 120, 7445–7448 CrossRef.
- (a) R. Brimblecombe, G. F. Swiegers, G. C. Dismukes and L. Spiccia, Inorg. Chem., 2009, 48, 7269–7279 CrossRef CAS; (b) R. Brimblecombe, A. M. Bond, G. C. Dismukes, G. F. Swiegers and L. Spiccia, Phys. Chem. Chem. Phys., 2009, 11, 6441–6449 RSC.
- G. C. Dismukes, R. Brimblecombe, G. A. N. Felton, R. S. Pryadun, J. E. Sheats, L. Spiccia and G. F. Swiegers, Acc. Chem. Res., 2009, 42, 1935–1943 CrossRef CAS.
- R. Brimblecombe, A. Koo, G. C. Dismukes, G. F. Swiegers and L. Spiccia, J. Am. Chem. Soc., 2010, 132, 2892–2894 CrossRef CAS.
- R. Brimblecombe, J. Chen, P. Wagner, T. Buchhorn, G. C. Dismukes, L. Spiccia and G. F. Swiegers, J. Mol. Catal. A, 2011, 338, 1–6 CAS.
- N. McCool, D. M. Robinson, J. E. Sheats and G. C. Dismukes, J. Am. Chem. Soc., 2011, 133, 11446–11449 CrossRef CAS.
- R. Hocking and L. Spiccia, Abstract 155, 39th International Conference on Coordination Chemistry, ICCC39, Adelaide, Australia, 2010 Search PubMed.
- J. E. Post and D. R. Verblen, Am. Mineral., 1990, 75, 477 CAS.
- K. Sauer and V. K. Yachandra, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 8631–8636 CrossRef CAS.
- D. M. Robinson, Y. B. Go, M. Greenblatt and G. C. Dismukes, J. Am. Chem. Soc., 2010, 132, 11467–11469 CrossRef CAS.
- J. C. Hunter, J. Solid State Chem., 1981, 39, 142–147 CrossRef CAS.
- Q. Yin, J. M. Tan, C. Besson, Y. V. Geletti, D. Musaev, A. E. Kuznetsov, Z. Luo, K. I. Hardcastle and C. L. Hill, Science, 2010, 328, 342–345 CrossRef CAS.
- See, for example: (a) X. Sala, I. Romero, M. Rodriguez, L. Escriche and A. Llobet, Angew. Chem., Int. Ed., 2009, 48, 2842–2852 CrossRef CAS; (b) M. Yagi and M. Kaneko, Chem. Rev., 2001, 101, 21–36 CrossRef CAS.
- C. F. Macrae, I. J. Bruno, J. A. Chisholm, P. R. Edgington, P. McCabe, E. Pidcock, L. Rodriguez-Monge, R. Taylor, J. van de Streek and P. A. Wood, J. Appl. Crystallogr., 2008, 41, 466–470 CrossRef CAS.
- (a) S. Petrie, R. Stranger and R. J. Pace, Angew. Chem., Int. Ed., 2010, 49, 4233–4236 CrossRef CAS , and references therein; (b) S. Petrie, R. Stranger and R. J. Pace, Chem.–Eur. J., 2010, 16, 14026–14042 CrossRef CAS.
- G. F. Swiegers in Mechanical Catalysis. Methods of Enzymatic, Homogeneous and Heterogeneous Catalysis, ed. G. F. Swiegers, John Wiley and Sons, New York, 2008, pp. 37–177 and 209–218 Search PubMed.
- See, for example: Y. Gao, T. Akermark, J. Liu, L. Sun and B. Akermark, J. Am. Chem. Soc., 2009, 131, 8726 CrossRef CAS , and references therein.
- C. Sun, Y. Wang, J. Zou and S. C. Smith, Phys. Chem. Chem. Phys., 2011, 13, 11325–11328 RSC.
- K. Kwon, K. Refson and G. Sposito, Phys. Rev. Lett., 2008, 100, 146601-1–146601-4 CrossRef.
- D. Quiñonero, A. L. Kaledin, A. E. Kuznetsov, Y. V. Geletii, C. Besson, C. L. Hill and D. G. Musaev, J. Phys. Chem. A, 2010, 114, 535–542 CrossRef.
- A. Sartorel, P. Miró, E. Salvadori, S. Romain, M. Carraro, G. Scorrano, M. Di Valentin, A. Llobet, C. Bo and M. Bonchio, J. Am. Chem. Soc., 2009, 131, 16051–16053 CrossRef CAS.
Footnote |
| † In this work we use the term “collision” in deference to Collision Theory;16,33 the correct term for liquid-phase reactions is an “encounter”. |
| This journal is © The Royal Society of Chemistry 2011 |