1H NMR metabolomics combined with gene expression analysis for the determination of major metabolic differences between subtypes of breast cell lines†
Miroslava
Cuperlovic-Culf
*ab,
Ian C.
Chute
c,
Adrian S.
Culf
bc,
Mohamed
Touaibia
d,
Anirban
Ghosh
c,
Steve
Griffiths
c,
Dan
Tulpan
a,
Serge
Léger
a,
Anissa
Belkaid
d,
Marc E.
Surette
d and
Rodney J.
Ouellette
cde
aNational Research Council of Canada, Institute for Information Technology, Moncton, Canada
bDepartment of Chemistry, Mount Allison University, Sackville, Canada
cAtlantic Cancer Research Institute, Moncton, Canada
dDépartement de Chimie et Biochimie, Université de Moncton, Moncton, Canada
eDépartement de Biochimie, Université de Sherbrooke, Sherbrooke, Canada
First published on 6th September 2011
Abstract
1H NMR analysis was performed on metabolic extracts from a selection of six breast cell lines, including normal-immortalized, invasive ductal carcinomas and adenocarcinomas. Metabolites with significant concentration differences between normal and cancerous cells as well as ER+ and ER− (estrogen receptor) cells were determined and their relation to the differentially expressed genes was explored. Major differences have been shown for many amino acids and this was linked to expression level changes of related genes. Observed changes in choline concentration were connected to expression level changes of the SCL44A1 transporter gene.
Introduction
Understanding how cancer cells derive energy and necessary building blocks, even from a nutrient depleted environment, is of fundamental importance for the development of appropriate therapies and diagnostic approaches.1–3 Tumourigenesis and progression is initiated, or at least accompanied, by a number of metabolic changes, some of which facilitate the processes of local invasion and metastasis to distant organs.4 The majority of cancers exhibit the “Warburg phenomenon” where cells demonstrate increased glycolysis leading to enhanced lactate production.1–3 In addition, many authors have reported changes in choline and phosphocholine (PCho) levels in cancers. For example, Aboagye and Bhujwalla5 demonstrated a gradual increase in both PCho levels and total choline-containing metabolite levels in breast cancer cells as they progress from normal to increasingly malignant phenotypes.Breast cancer is a biologically heterogeneous disease with demonstrated differences in gene expression profiles6 and clinical outcomes. Breast cancer subtypes appear to also possess different metabolic characteristics. Recently, in vivo studies have revealed differences in choline levels between subtypes7 whereas in vitro studies of cells have shown that ER+ cells with low metastatic potential have higher levels of PCho, phosphodiesters and uridine diphosphosugar than the ER− cells.8,9 However, other metabolic changes in breast cancers and their subtypes remain to be elucidated.
High throughput metabolite profiling (metabolomics) strategies provide an excellent approach for un-biased analysis of metabolic differences in cancer subtypes. Metabolomics is particularly important for the search for biomarkers that can be eventually used for in vivo diagnosis and prognosis through methods such as magnetic resonance spectroscopy (MRS). The first step towards devising a metabolite based non-invasive breast cancer subtyping method is to obtain an accurate representation of major, MRS visible, metabolic differences and markers in cancer subtypes. An initial effort was undertaken by the parallel analysis of tumour samples from 46 breast cancer patients.10 Borgan and co-workers performed the measurement of metabolic profiles by NMR and gene expression profiles by microarrays on the same tissues resulting in a correlative relationship between these two datasets. However, although in principle analysis of tissues is of major interest, a complex heterogeneity of tumour tissues and the varying presence of non-tumour cells in the samples render the interpretation of the results problematic (average containment of tumour cells in the samples was reported to be 23% with a range 0–80% across the samples10). Thus, we believe it is appropriate to first identify metabolic profiles in a controlled setting such as breast cancer cell lines as a prerequisite to the analysis of a more complex case of the entire tumour tissues or biopsies. The work of Neve et al.11 has shown that breast cell lines display the same genetic (heterogeneity in copy number) and gene expression abnormalities as the primary tumours from which they were derived. Furthermore, cell line collections exhibit similar responses to targeted therapeutics as observed in clinics.11 With the observed correlation in transcription profiles, it can be expected that metabolic measurements of cancer cell lines would provide a sound model for primary tumours without heterogeneities and confounding factors that are generally unavoidable in clinical samples.
In this work we are exploring specific metabolic characteristics of different breast cell lines with the ultimate goal of determining differences between breast tumour subtypes. We have analyzed metabolic profiles of six different breast cell lines that represent immortalized normal, as well as invasive and non-invasive tumour cell lines with both ER+ and ER− backgrounds. These metabolic profiles are compared with publicly available gene expression data in order to determine any relationships between metabolic and gene expression alterations.
Results and discussion
Metabolite analysis was performed on six different breast cell lines including two normal-immortalized cell lines (MCF10A and MCF12A), two invasive ductal carcinomas (non-metastatic MCF7 and metastatic T47D) and two adenocarcinomas (MB231 and SKBR3). These cell lines, with different ER, PR, HER2 and TP53 characteristics, provide a good representation of a variety of breast cell lines and breast cancers. The most prominent characteristics of cell types included in this work are provided in Table 1.11| Cell line | Type | Gene cluster | ER, PR, HER2 | Culture mediuma |
|---|---|---|---|---|
| a All cell lines were cultured in glucose reduced DMEM for 24 h prior to harvesting. | ||||
| MCF10A | Fibrocystic disease | Basal B | −, −, − | DMEM/F12, 5% FBS |
| MCF12A | Fibrocystic disease | Basal B | −, −, − | DMEM/F12, 5% FBS |
| MCF7 | Invasive ductal carcinoma (IDC) | Luminal | +, +, − | DMEM, 10% FBS |
| MB231 | Adenocarcinoma (AC) | Basal B | −, −, − | DMEM, 10% FBS |
| SKBR3 | Adenocarcinoma (AC) | Luminal | −, −, + | McCoys 5A, 10% FBS |
| T47D | Invasive ductal carcinoma (IDC) | Luminal | +, +, − | RPMI, 10% FBS |
Five independent (biological) replicates of cells were grown in optimal culture media (Table 1 and Materials and methods) until the 24 h prior to harvesting when all cells received the same medium. These treatments ensured that cells had been grown under their optimal conditions, but prior to harvesting the media of the cell lines were equalized in order to reduce the influence of different culture media on results.
The metabolites were extracted for each replicate of each of the 6 cell lines separately using the procedure based on the work of Dietmair et al.15 (see Materials and methods). 1D 1H NMR spectra for 30 metabolic extracts were measured and used for analysis. Additionally, 1D 13C and 2D TOCSY and HMQC spectra were obtained for a pool of all six cell types and were used for metabolite assignment.
The mean NMR spectra obtained for each cell line type are shown in Fig. 1. The individual spectra for all replicates for each cell line are available upon request. Spectra of biological replicates show negligible differences. The analysis of metabolic profiles was performed qualitatively, directly on the spectra, as well as semi-quantitatively, on quantified peak values. Sample spectra and the peak intensities were analyzed with unsupervised, clustering methods in order to determine the similarities and differences between the sample types in an unbiased fashion. Next, both qualitative and quantitative data were analyzed with supervised feature selection methods in order to determine specific differences between sample types. The major differential features were assigned to metabolites and these assignments were explored in relation to gene expression data. (Methods are described in detail in the ESI.†)
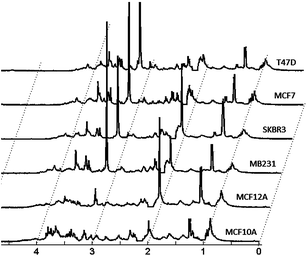 | ||
| Fig. 1 Average 1D 1H NMR spectra of 5 biological replicates for 6 cell lines. Cell lines included in the study are described in Table 1. Only the spectral region between 0 and 4.5 ppm is shown. Spectral points in the region between 2.1 and 2.2 ppm contain residual hydrogen-containing solvent and are therefore removed. | ||
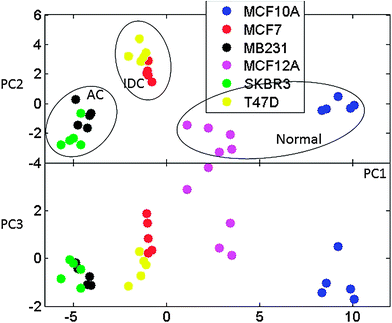
In the first round of analysis the complete spectra were binned into different bin sizes and normalized using the total peak area normalization method. The qualitative analysis of the major variances in the spectra was performed directly by using principal component analysis (PCA) as well as by clustering samples with the fuzzy K-means method (FCM). The results are presented in Fig. 2 and 3.
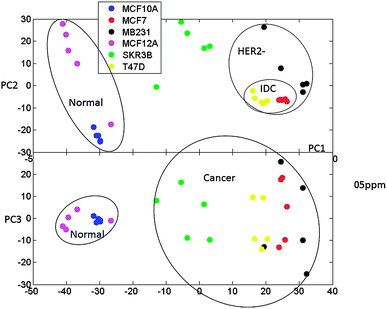 | ||
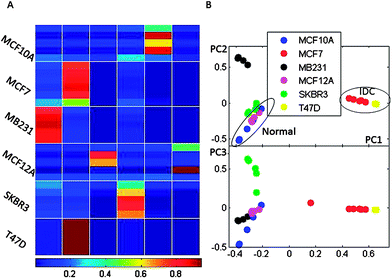
| Fig. 2 Principal components PC1 vs. PC2 and PC1 vs. PC3 for PCA analysis of spectral data for six cell line types. Metabolites were independently extracted and measured for five biological replicates corresponding to six cell line types. The grouping of normal and cancer as well as IDC− and HER− cell types is outlined. For the presented analysis, spectra were binned to 0.005 ppm sized segments. | ||
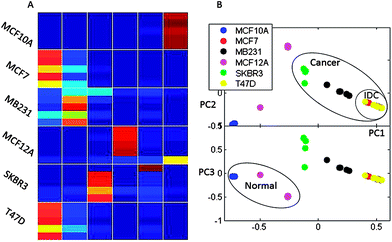 | ||
| Fig. 3 FCM clustering of binned spectral data. Presented are: A. The membership values for each measurement where red represents the membership value of 1 and dark blue corresponds to a membership of 0. Higher membership values indicate stronger belonging to a cluster. B. Principal components analysis of membership values. PCA analysis is performed for visualization of FCM results and shows major groups of samples based on the membership values obtained by FCM. Cancer, normal and IDC groups are indicated on the PCA plot. | ||
Fig. 2 shows the plot of principal components PC1, PC2 and PC3 for the spectral data binned to 0.005 ppm for the five replicates of the six cell lines studied. Similar separation of samples with PCA has been obtained with different bin sizes ranging from 0.005 to 0.01 ppm.
Fig. 3 shows the membership values (Fig. 3A) for fuzzy clustering as well as the PCA of membership values for visualization of the results (Fig. 3B). The two analysis methods show separation of normal and cancer cells. The samples belonging to the two normal cell types are assigned to different clusters with the FCM method. This can be ascribed to differences between the sources of these two normal cell cultures (MCF12A cells are obtained from a 60 year patient and MCF10A cells from a 36 year old patient with fibrocystic disease). Further analysis of membership values (Fig. 3B, PC3 vs. PC1 plot) shows similarity between the two normal cell lines in comparison to the four cancer cell lines. Concordantly, within the cancer cell types, IDC cells (MCF7 and T47D) appear to be more metabolically related to each other than to the AC cells (SKBR3 and MB231). Therefore, as the ER+ cells (MCF7 and T47D) are clearly co-clustering based only on a qualitative analysis of metabolic profiles, we can hypothesize that the ER status has a significant influence on metabolic profiles. The determination of specific metabolites that are most significantly different between ER+ and ER− cells is a later focus in this manuscript. Further, HER2− cells (MCF7, MB231 and T47D) are once again more closely grouped. The SKBR3 cells, which are ER−/PR−/HER2+, are separately clustered from all the other cancer lines. Although SKBR3 is more closely grouped with MB231 cells (ER−/PR−/HER2−), it is clear from FCM membership values that the MB231 metabolic profile has more similarity with IDC lines (ER+/PR+/HER2−) than the SKBR3 line. From these results it can be hypothesized that according to the NMR metabolic profiles HER2 expression also has an influence on cellular metabolic profile.
Semi-quantitative analysis used the peak areas in the spectra that were determined by the global spectra deconvolution method (GSD) and total lineshape analysis (TLS). PCA, clustering and major differential features determination was possible with the peak intensities dataset as well. The PCA and FCM analyses of GSD peaks are presented in Fig. 4 and 5. The cluster analysis of TLS data provides a comparable result (data not shown).
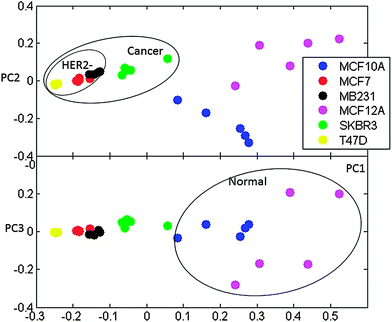 | ||
| Fig. 4 Principal components PC1 vs. PC2 and PC1 vs. PC3 for PCA analysis of GSD peak values for spectra. The separation of normal and cancer as well as IDC and HER− cell types is outlined. | ||
 | ||
| Fig. 5 FCM clustering of GSD data. Presented are: A. The membership values for each measurement where red is a membership value of 1 and dark blue shows a membership of 0. Higher membership values indicate stronger belonging to a cluster. B. Principal components derived from a PCA analysis of membership values. PCA analysis is performed for visualization of FCM results and shows major groups of samples based on the membership values obtained by FCM. Normal and IDC groups are indicated on the PCA plot. | ||
The final GSD dataset included 79 peaks. Generally, PCA and FCM analysis of GSD peaks (as well as TLS peaks) provides comparable results to the qualitative analysis of binned spectra. Once again cancer lines are clearly separated. The similarity of metabolic profiles of IDC cell lines is even more apparent in this analysis with MCF7 and T47D lines clearly co-clustering. Although PCA plots cannot clearly separate MCF7 and MB231 lines, FCM clustering very clearly separates these two subtypes of cancer while co-clustering the two IDC lines (MCF7 and T47D).
It is evident from these data that the unsupervised analysis of qualitative and quantitative NMR metabolomics profiles leads to similar conclusions. Clearly, and not surprisingly, normal and cancer cell lines have different metabolic profiles. Many studies have previously shown differences in energy metabolism (with changes particularly in glycolysis and TCA pathways) as well as other pathways including amino acid production pathways, lipid processing and synthesis pathways, etc. Furthermore, unsupervised analysis presented here shows the dissimilarities in metabolic profiles of cancer subtypes particularly between ER+ invasive ductal carcinomas (IDC) and ER− adenocarcinomas (AC).
In order to determine specific spectral features and ultimately metabolites that have the most significant concentration difference between normal and cancer and between AC and IDC phenotypes, we have performed supervised feature selection using the SAM method13 included in the TMeV gene expression analysis software package.22
Although both SAM and TMeV were originally developed for the analysis of gene expression data, these tools are universal data analysis methods that can be used for both qualitative, i.e. spectral, or quantitative, i.e. peak, metabolomics data. With SAM we have selected major differential features between normal (MCF10A and MCF12A) and cancer (MB231, MCF7, SKBR3 and T47D) cell lines as well as between IDC (MCF7 and T47D) and AC (MB231 and SKBR3) cancer cell lines.
Although there are small discrepancies in some major feature positions obtained with two qualitative and semi-quantitative methods, in general the analysis of spectral and GSD data is in agreement (the two lists of features are shown relative to the 1D data in the ESI†). The spectral analysis leads to a larger number of significant features than the equivalent analysis of GSD data. This is not surprising considering that spectral data generally have more than one point for each peak. Furthermore, in the crowded spectra obtained here, optimized GSD peak analysis can miss overlapping features. Therefore, we have used both major features that were obtained from spectral and from GSD data for the determination of changed metabolites in different groups of samples. Metabolite assignment was based on 1D 1H, TOCSY, HMQC and 13C measurements. Major features and assignments are shown relative to TOCSY results in Fig. 6 and 7, with obtained metabolites and their relative levels in the studied groups provided in Table 2. Major features are also outlined on 1D and HMQC spectra in Supplementary Fig. 1–4.† Assignments are based on previously published data18 and the automatic assignment method developed in our group. This method, called MetaboHunter is based on the advanced search of peaks in the spectra using the HMDB23 and Madison spectral libraries. The MetaboHunter method will be explained in more detail in different publication (in preparation). In addition, the list of metabolites obtained from these assignment methods was compared with metabolites that were identified through HPLC separation by Dietmair et al.15 Only those metabolites that were previously observed in acetonitrile extracts were included in our final list of significantly different metabolites for the two comparisons (Table 2). This relatively short list of metabolites provides information about the major differences observable by NMR for further comparison with gene expression measurements.
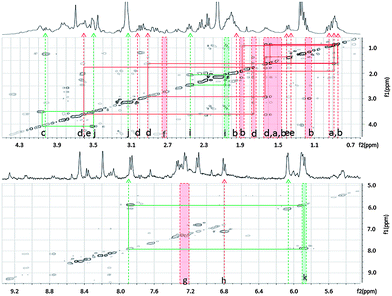 | ||
| Fig. 6 Major differential features between cancer and normal cell types determined with SAM. Indicated in the figure are significantly higher intensity features in cancer cell types (green) and in normal cell types (red). The major features are shown against a TOCSY experiment background. Metabolite assignments for these major significantly different features are listed in Table 2A. | ||
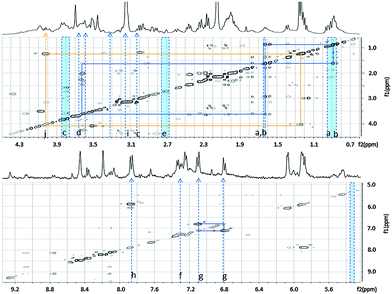 | ||
| Fig. 7 Major differential features between IDC and AC cell types obtained using SAM. Indicated are features with significantly higher intensity in IDC lines (blue) and in AC cell lines (orange). The major features are represented on a TOCSY experiment background. Metabolite assignments are listed in Table 2B. | ||
| A | Metabolite | ppm range | Cancer average | Normal average |
|---|---|---|---|---|
| a | L-Valine | 0.75–1.00 | 73.01(1.36) | 110.53(9.12) |
| b | L-Leucine | |||
| L-Isoleucine | ||||
| d | L-Lysine | 1.675–1.78 | 10.31(1.36) | 20.55(2.21) |
| e | L-Alanine | 1.35–1.40 | 11.62(1.54) | 16.35(1.1) |
| f | L-Aspartic acid | 2.73–2.745 | 0.77(0.08) | 2.65(0.71) |
| g | Phenylalanine | 7.21–7.35 | 6.06(0.96) | 11.12(1.88) |
| h | Tyrosine | 6.99–7.01 | 0.25(0.12) | 0.50(0.14) |
| i | Glutamine | 2.44–2.46 | 5.53(0.83) | 3.44(0.81) |
| j | Total choline | 3.15–3.16 | 13.93(5.32) | 6.58(1.84) |
| k | UDP-glucose | 5.835–5.95 | 6.59(0.75) | 1.63(1.63) |
| c | Lactic acid | 1.22–1.27 | 58.29(13.84) | 56.47(18.19) |
| B | Metabolite | ppm range | IDC (ER+) average | AC (ER−) average |
|---|---|---|---|---|
| a | L-Valine | 0.75–1.00 | 78.05(1.76) | 67.98(6.10) |
| b | L-Leucine | |||
| L-Isoleucine | ||||
| c | Glycerol-3-phosphate | 3.72–3.88 | 50.31(4.56) | 38.27(2.12) |
| d | L-Alanine | 1.35–1.40 | 12.06(0.59) | 11.17(2.05) |
| e | L-Aspartic acid | 2.73–2.745 | 0.79(0.07) | 0.74(0.09) |
| f | Phenylalanine | 7.21–7.35 | 6.82(0.62) | 5.23(0.51) |
| g | Tyrosine | 6.99–7.01 | 0.26(0.08) | 0.24(0.16) |
| i | Choline | 3.15–3.16 | 15.02(5.01) | 12.84(5.66) |
| j | Lactic acid | 1.22–1.27 | 49.2(3.19) | 67.39(14.51) |
Many of the metabolites obtained as significant in comparison between tumour and normal cell lines have previously been reported. A large body of research has already established that there are higher concentrations of choline and lactic acid in cancers. Choline is part of several pathways, most importantly metabolism of glycerophospholipids and other lipids. The analysis of the glycerophospholipid pathway shows over-expression of several important enzymes related to choline processing as well as genes involved in choline import into cells. The lactic acid up regulation in cancer has also been observed and can be ascribed to increased glycolysis (Warburg's effect) and a reduced TCA (citric acid) cycle in cancers. The increased concentration of glutamine in cancers has been reviewed by Dang.16 It was proposed that glutamine is transported into the cell through ASCT2 and converted to glutamate by glutaminase (GLS), which is under the control of MYC. Glutamate is further catabolized to α-ketoglutarate (α-KG) for further oxidation in the TCA cycle. Malate generated from α-ketoglutarate can exit the TCA cycle for conversion to pyruvate and ultimately lactic acid in the cytoplasm. UDP-glucose is present in several pathways including glycogen metabolism, N-glycan biosynthesis and sphingolipid metabolism, all of which have been previously related to the cancer metabolic phenotype. In Fig. 8 we show the connection between over-concentrated metabolites in cancers: L-glutamine, L-lactate, choline and genes with expression measured as part of the work of Ross and Perou.6 Clearly there are two transporter genes: SLC3A2 and SCL44A1 that are over-expressed in cancer and that have been previously related to the transport of L-glutamine and choline. SLC3A2 has been proposed as a mediator of arginine, leucine and glutamine uptake.26 SCL44A1 (intermediate-affinity choline transporter-like protein) has been indicated as a major choline transporter in breast cancers with a connection to the Hsp90 chaperon protein.29 At the same time, epidermal growth factor receptor (EGFR) which is under-expressed in cancer has been indicated as part of the mechanism for the reduction of monolayer permeability to choline.27 The transporters related to lactate are under-expressed in cancer, clearly indicating that lactate is produced in the cell rather than imported (in this population of cancer cells). This is in clear agreement with the over-expression of enzymes involved in the production of lactate from glutamine (through a changed TCA cycle) as well as through glycolysis.
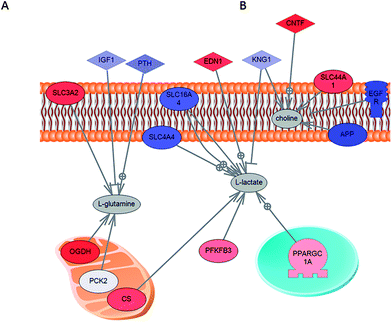 | ||
| Fig. 8 Direct connection network between gene expression and metabolites over-concentrated in cancer cells. Genes that are directly involved in the transport of L-glutamine and choline (SLC3A2 and SCL44A1) are over-expressed. L-Lactate is clearly not imported into cells (transporters are under-expressed in this case) but is produced as part of the cancer glycolysis pathway or from L-glutamine (with over-expression of related enzymes: OGDH and CS). In the figure genes under-expressed in cancers are shown in blue and in red are genes that are over-expressed in cancers. | ||
All metabolites that were determined with these methods to be under-concentrated in cancers are amino acids. According to this study, the amino acids with reduced concentrations in cancer cells include lysine, isoleucine, aspartic acid, valine, alanine, phenylalanine, tyrosine and leucine. It is very interesting to observe that these include the branched-chain amino acids BCAAs (arginine, valine, leucine and isoleucine). BCAAs are used by cells for protein synthesis or are catabolised into sources for glucose and lipid production. These branched-chain amino acids influence proteolysis, hormone release and cell cycle progression along with their other metabolic roles. The branched-chain amino acids play a central role in regulating cellular protein turnover by reducing autophagy,19 all of which are significant in the development and progression of cancer. Furthermore, previous work indicated a possible role of BCAAs in inducing apoptosis.21 It has been proposed that amino acids necessary for cancer cell development result from glutaminolysis and changed TCA cycle function.17 In this model BCAAs are immediately used, thus leading to a reduction of free amino acid concentrations particularly in the case of nutrient starvation. The exploration of the branched chain amino acids metabolism pathway shows that the majority of enzymes in this pathway are over-expressed in cancers (data not shown) thus leading to a faster inclusion of these amino acids in proteins and therefore reduced concentrations of free amino acids. A similar result is observed with the alanine pathway that includes alanine and aspartate. In this case the branch of the pathway that is depleting alanine and aspartate contains over-expressed genes.
Estrogens are important regulators of growth and differentiation in the normal mammary gland and participate in the development and progression of breast cancer.14,24,25 The effect of estrogens on breast cells results from the estrogen receptor mediated regulation of expression of a range of genes including those involved in cell cycle regulation. The changes in gene expression provoked by the estrogen receptor can also lead to alterations in metabolic profiles. Previous research has shown that the introduction of ERα into the ER− cell line (MB231) alters expression of many genes, including several enzymes that are involved in the metabolism of fatty acids, carbohydrates, nucleic acids and amino acids as well as glycolysis and glycerolipid metabolism and folate biosynthesis.14,25 The metabolic changes that were observed in the present study indicate related alterations in metabolites between ER+ and ER− cell lines. NMR profiles of ER+ and ER− cells indicate changes in several metabolites including lactic acid, which is present at lower concentrations in ER+ cancers, as well as valine, leucine, isoleucine, alanine, aspartic acid, phenylalanine, tyrosine, glycerol-3-phosphate and choline, which are more concentrated in ER+ cancers. In terms of amino acid metabolism, ER+ cells appear to be metabolically closer to normal cells than are ER− cell lines. Furthermore, with a higher concentration of lactic acid in ER− cells, it appears that the Warburg effect is more significant in these cases. The work of Obayashi et al.20 has shown estrogen control of BCAA catabolism. The difference in concentrations of BCAAs between ER+ and ER− cell lines observed here is clearly in agreement with Obayashi's results. Furthermore, as in the case of the cancer/normal analysis, in a ER+/ER− comparison those genes involved in BCAA catabolism are over-expressed in ER− cell lines, likely leading to the depletion of BCCAs. Total choline is determined to be more concentrated in ER+ cell lines. This can be easily understood when observing the expression pattern of choline transport related genes: SLC44A1 and EGFR (Fig. 9). It is clear from the figure that the expression levels of these two genes correspond to increased input of choline into cells, as was observed with the comparison between cancer and normal cell types. Furthermore, previous analysis specifically focused on choline derivatives has shown a higher concentration of phosphocholine in ER+ breast tissue samples.28 This concentration difference is in correlation with genes related to phosphocholine synthesis.30 Moreover, glycerol-3-phosphate, which is also significantly over-expressed in IDC cells, is a key intermediate in the metabolism of glycerophospholipids into choline.
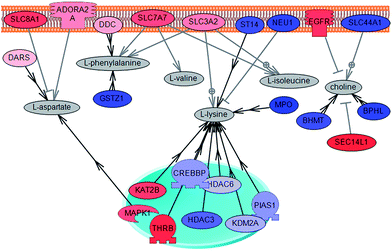 | ||
| Fig. 9 Direct connection network between metabolites over-concentrated in ER+ relative to ER− cells. In the figure the genes that are over-expressed in ER+ cells are shown in blue and presented in red are genes that are over-expressed in ER− cells. Gene SLC44A1, involved in transport of choline, is over-expressed in ER+ cells. | ||
It is interesting to also point out that the metabolic differences observed between AC and IDC cells relate to differences between normal and cancer cell lines. In fact, PCA of spectral data for SAM determined most significantly different intensities in the spectra (defined as spectral positions in ppm) between IDC and AC cells (Fig. 10) shows the continuous change from normal, via IDC and to AC cell types. Therefore, it is conceivable that detailed quantitative measurements of obtained metabolic markers can be used for both diagnosis and type determination in breast cancers.
 | ||
| Fig. 10 Principal components PC1 vs. PC2 and PC1 vs. PC3 for PCA analysis of SAM determined major features differentiating between AC and IDC cell lines. The separation of normal, IDC and AC cell types is indicated. | ||
Conclusions
We have performed unsupervised and supervised analysis of 1D 1H NMR measurements of six different breast cell lines. The presented analysis shows that normal and cancer cell lines as well as cell lines of different cancer subtypes can be separated based on their metabolic profiles. In addition to unsupervised separation of cell types, we have determined the major differentially regulated metabolites between cancer and normal cells and between ER+ and ER− cancer cells. Metabolic analysis generally agrees with gene expression measurements for these cell lines. The presented work points to the major metabolic differences between breast cell lines that represent different breast cell types. We believe that this is an important step towards the determination of metabolic markers for breast cancer type identification through the non-invasive, in vivo MRS of tissues. Furthermore, the SCL44A1 gene appears to be significant in choline transport and in the differences observed in choline transport in breast cancer subtypes and, therefore, presents an interesting target for future analysis. Future work will include the application of different metabolite extraction protocols for metabolite analysis and assignment as well as quantification of a larger number of metabolites and samples.Materials and methods
Cell lines and in vitro culture conditions
All cell lines were obtained from ATCC (Manassas, VA, USA). All media and components were purchased from Invitrogen unless otherwise noted. MCF10A and MCF12A cells were grown in Dulbecco's modified Eagle's medium/Ham's F12 (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v/v) supplemented with 2 mM L-glutamine, 1 mM sodium pyruvate, 20 ng mL−1 epidermal growth factor (Sigma Aldrich), 100 ng mL−1 cholera toxin (Sigma Aldrich), 0.01 mg mL−1 bovine insulin (Sigma Aldrich), 500 ng mL−1 hydrocortisone (Sigma Aldrich), 5% fetal bovine serum (PAA) and penicillin/streptomycin (100 U mL−1 and 100 μg mL−1, respectively). MDA-MB-231 and MCF7 cells were grown in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum (PAA), 2 mM L-glutamine and penicillin/streptomycin. SKBR3 cells were grown in McCoy's 5A medium supplemented with 10% fetal bovine serum (PAA) and penicillin/streptomycin. T47D cells were grown in RPMI 1640 medium supplemented with 10% fetal bovine serum (PAA) and penicillin/streptomycin.
:1, v/v) supplemented with 2 mM L-glutamine, 1 mM sodium pyruvate, 20 ng mL−1 epidermal growth factor (Sigma Aldrich), 100 ng mL−1 cholera toxin (Sigma Aldrich), 0.01 mg mL−1 bovine insulin (Sigma Aldrich), 500 ng mL−1 hydrocortisone (Sigma Aldrich), 5% fetal bovine serum (PAA) and penicillin/streptomycin (100 U mL−1 and 100 μg mL−1, respectively). MDA-MB-231 and MCF7 cells were grown in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum (PAA), 2 mM L-glutamine and penicillin/streptomycin. SKBR3 cells were grown in McCoy's 5A medium supplemented with 10% fetal bovine serum (PAA) and penicillin/streptomycin. T47D cells were grown in RPMI 1640 medium supplemented with 10% fetal bovine serum (PAA) and penicillin/streptomycin.
10 cm culture-treated dishes (5 replicates per cell line) were seeded with approximately 3 × 106 cells and incubated for 24 h at 37 °C and 5% CO2. Following this period, the seeding media for all cell types were removed and replaced with FBS-free Dulbecco's modified Eagle's medium. The cells were then incubated for a further 24 h prior to harvest. Cells were harvested by scraping and washed with PBS before being spun down by centrifugation at 4000 RCF (relative centrifugal force) for one minute. Cell pellets were kept on ice for 5 min before being re-suspended in 1 mL of 50% acetonitrile (Fisher Scientific). Cell suspensions were kept on ice for 10 min before being spun at 16000 RCF for 10 min at 4 °C.
NMR experimentation
The aqueous acetonitrile extract solution was dried at 50 °C by rotary evaporation for 3 min. The white residue was dissolved in 0.6 mL of deuterium oxide (Aldrich, 99.96 atom% 2H), pipetted into a glass 5 mm NMR tube and sealed with Parafilm for NMR analysis.All 1H NMR measurements were performed on a Bruker Avance III 400 MHz spectrometer. 1D spectra were obtained using a gradient water presaturation method with 512 scans. NMR spectra were processed using Mnova with exponential apodization (exp 1); global phase correction; Berstein polynomial baseline correction; Savitzky–Golay line smoothing and normalization using total spectral area as provided in Mnova. Spectral regions from 0–2.1 ppm and 2.2–9 ppm were included in the normalization and analysis. 1D 13C and 2D spectra, including TOCSY and HMQC using standard methods provided in TopSpin, Bruker, were performed on a pool of metabolic extracts from all six cell lines. Metabolite assignment was performed using 2D measurements as well as 1D 1H and 13C spectral data for the regions that were determined as significantly different in analysed groups. Only metabolites that could be shown present on both 1D and 2D spectra were assigned.
Data analysis
Data pre-processing including data organization, removal of undesired areas and binning as well as data presentation was performed with MATLAB vR2010b. PCA as well as fuzzy K-means cluster analysis were also performed under the MATLAB platform as described previously.12 Feature selection was done with the significance analysis of microarrays (SAM) method.13 A more detailed description of these two methods is provided in the ESI.† Automatic peak assignment was performed using several methods developed in our group and elsewhere (HMDB, http://www.hmdb.ca). Spectra for suggested differentially present metabolites are obtained from the Human Metabolomics Database (http://www.hmdb.ca) and further analysed visually and compared to the obtained spectra.23The peak areas in the spectra were determined using the global spectra deconvolution method (GSD) as provided in Mnova and the total lineshape analysis (TLS) provided by Perch Solutions. Briefly, this method automatically reduces a frequency domain spectrum to a set of Lorentzian or near-Lorentzian lines leaving out any baseline drift and noise. GSD and TLS automatically deconvolute all the peaks in a spectrum and provide information about the optimized list of peaks with their positions and intensities for each spectrum. GSD automatically deconvolutes all the peaks in a spectrum by first recognizing all significant peaks, followed by assigning realistic a priori bounds to all peak parameters (chemical shift, heights, line widths, etc.) and finally fitting all these parameters. The output of GSD is a “peak list” of spectral parameters for Lorentzian lines in terms of frequency, amplitude, line width and, optionally, phase of all the desirable information present in the original spectrum but none of the superfluous “noise”. These peaks can then be subjected to automatic and/or manual editing such as automatic recognition of spikes (anomalously narrow peaks), solid impurities (very broad peaks), folded-over peaks (anomalous phase), rotation sidebands and isotopomer satellites. GSD peak positions and intensities for each spectrum were combined using in-house developed MATLAB (MathWorks) routines. Only GSD peaks that were determined in more than 80% of all samples were used in the analysis. The missing peaks were assigned an intensity of zero and were not subsequently considered.
Microarray data
Two microarray datasets used in this work were previously validated for accuracy in RNA expression measurements. The first set provided by Neve et al.11 includes Affymetrix microarray measurements for many different cell lines and primary tissues. A second dataset, provided by Ross and Perou6includes spotted microarray data also for many different breast cell lines. Both datasets included microarray measurements for the six cell lines studied here. The descriptions of methods used for sample processing, microarray experimentation and validation are given in the original publications. These microarray data were analysed using TMeV22 and Pathway Studio (Ariadne Inc.). TMeV is the general data analysis software which includes many features such as normalization, clustering and statistical analysis. Pathway Studio is the commercial software which determines relationships between biological molecules and terms based on extensive and highly advanced literature searches.Acknowledgements
We would like to acknowledge the contributions from the Canada Foundation for Innovation (CFI), the New Brunswick Innovation Fund (NBIF) and the Université de Moncton for the acquisition of the NMR instrument. MES is supported by the Canada Research Chairs program. MC and MT would like to thank M. Monette (Bruker Canada) for her help in setting up NMR experiments.Notes and references
- G. Kroemer, Cancer Cell, 2008, 13, 472 CrossRef CAS.
- M. C. Brahimi-Horn, J. Chiche and J. Pouyssegur, Curr. Opin. Cell Biol., 2007, 19, 223 CrossRef CAS.
- O. Warburg, K. Posener and E. Negelein, Biochem. Z., 1924, 152, 319 Search PubMed.
- A. M. Weljie, A. Bondareva, P. Zang and F. R. Jiri, J. Biomol. NMR, 2011, 49, 185 CrossRef CAS.
- E. O. Aboagye and Z. M. Bhujwalla, Cancer Res., 1999, 59, 80 CAS.
- D. T. Ross and C. M. Perou, Disease Markers, 2001, 17, 99 CAS.
- H. M. Baek, J. H. Chen, K. Nie, H. J. Yu, S. Bahri, R. S. Mehta, O. Nalcioglu and M. Y. Su, Radiology, 2009, 251, 653 CrossRef.
- C. Oakman, L. Tenori, L. Biganzoli, L. Santarpia, S. Cappadona, C. Luchinat and A. Di Leo, Int. J. Biochem. Cell Biol., 2011, 43, 1010 CrossRef CAS.
- M. Sterin, J. S. Cohen, Y. Mardor, E. Berman and I. Ringel, Cancer Res., 2001, 61, 7536 CAS.
- E. Borgan, B. Sitter, O. C. Lingjærde, H. Johnsen, S. Lundgren, T. F. Bathen, T. Sørlie, A. L. Børresen-Dale and I. S. Gribbestad, BMC Cancer, 2010, 10, 628 CrossRef CAS.
- R. M. Neve, K. Chin, J. Fridlyand, J. Yeh, F. L. Baehner, T. Fevr, L. Clark, N. Bayani, J. P. Coppe, F. Tong, T. Speed, P. T. Spellman, S. DeVries, A. Lapuk, N. J. Wang, W. L. Kuo, J. L. Stilwell, D. Pinkel, D. G. Albertson, F. M. Waldman, F. McCormick, R. B. Dickson, M. D. Johnson, M. Lippman, S. Ethier, A. Gazdar and J. W. Gray, Cancer Cell, 2006, 10, 515 CrossRef CAS.
- M. Cuperlovic-Culf, N. Belacel, A. S. Culf, I. C. Chute, R. J. Ouellette, I. W. Burton, T. K. Karakach and J. A. Walter, Magn. Reson. Chem., 2009, 47, S96 CrossRef CAS.
- V. G. Tusher, R. Tibshirani and G. Chu, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 5116 CrossRef CAS.
- J. G. Moggs, T. C. Murphy, F. L. Lim, D. J. Moore, R. Stuckey, K. Antrobus, I. Kimber and G. Orphanides, J. Mol. Endocrinol., 2005, 34, 535 CrossRef CAS.
- S. Dietmair, N. E. Timmins, P. P. Gray, L. K. Nielsen and J. O. Kromer, Anal. Biochem., 2010, 404, 155 CrossRef CAS.
- C. V. Dang, Cancer Res., 2010, 70, 859 CrossRef CAS.
- R. A. Cairns, I. S. Harris and T. W. Mak, Nat. Rev. Cancer, 2011, 11, 85 CrossRef CAS.
- K. E. Cano, Y. Li and Y. Chen, J. Proteome Res., 2010, 9, 5382 CrossRef CAS.
- C. B. Doering and D. J. Danner, Cell Physiol., 2000, 279, C1587 CAS.
- M. Obayashi, Y. Shimomura, N. Nakai, N. H. Jeoung, M. Nagasaki, T. Murakami, Y. Sato and R. A. Harris, J. Nutr., 2004, 134, 2628 CAS.
- P. Jouvet, P. Rustin, D. L. Taylor, J. M. Pocock, U. Felderhoff-Mueser, N. D. Mazarakis, C. Sarraf, U. Joashi, M. Kozma, K. Greenwood, A. D. Edwards and H. Mehmet, Mol. Biol. Cell, 2000, 11, 1919 CAS.
- E. Howe, K. Holton, S. Nair, D. Schlauch, R. Sinha and J. Quackenbush, Biomedical Informatics for Cancer Research, 2010, 267 CrossRef.
- D. S. Wishart, C. Knox, A. C. Guo, R. Eisner, N. Young, B. Gautam, D. D. Hau, N. Psychogios, E. Dong, S. Bouatra, R. Mandal, I. Sinelnikov, J. Xia, L. Jia, J. A. Cruz, E. Lim, C. A. Sobsey, S. Shrivastava, P. Huang, P. Liu, L. Fang, J. Peng, R. Fradette, D. Cheng, D. Tzur, M. Clements, A. Lewis, A. De Souza, A. Zuniga, M. Dawe, Y. Xiong, D. Clive, R. Greiner, A. Nazyrova, R. Shaykhutdinov, L. Li, H. J. Vogel and I. Forsythe, Nucleic Acids Res., 2009, 37(Database), D603 CrossRef CAS.
- M. C. Pike, D. V. Spicer, L. Dahmoush and M. F. Press, Epidemiol. Rev., 1993, 15, 17 CAS.
- J. A. Vendrell, F. Magnino, E. Danis, M. J. Duchesne, S. Pinloche, M. Pons, D. Birnbaum, S. Nguyen, C. Theillet and P. A. Cohen, J. Mol. Endocrinol., 2004, 32, 397 CrossRef CAS.
- A. Bröer, C. A. Wagner, F. Lang and S. Bröer, Biochem. J., 2000, 349, 787 Search PubMed.
- Y. Peter, A. Comellas, E. Levantini, E. P. Ingenito and S. D. Shapiro, Mol. Carcinog., 2009, 48, 488 CrossRef CAS.
- S. A. Moestue, E. Borgan, E. M. Huuse, E. M. Lindholm, B. Sitter, A. L. Børresen-Dale, O. Engebraaten, G. M. Maelandsmo and I. S. Gribbestad, BMC Cancer, 2010, 10, 433 CrossRef CAS.
- A. H. Brandes, C. S. Ward and S. M. Ronen, Breast Cancer Res., 2010, 12, R84 CrossRef.
- D. L. Morse, D. Carroll, S. Day, H. Gray, P. Sadarangani, S. Murthi, C. Job, B. Baggett, N. Raghunand and R. J. Gillies, NMR Biomed., 2009, 22, 114–127 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Fuzzy K means clustering methodology, significant analysis of microarrays and supplementary figures. See DOI: 10.1039/c1sc00382h |
| This journal is © The Royal Society of Chemistry 2011 |