Opening the silole ring: Efficient and specific cleavage of the endo-C(sp2)-Si bond with AcOH/ROH system†
Qian
Luo
a,
Chao
Wang
bc,
Yuexing
Li
a,
Kunbing
Ouyang
a,
Li
Gu
a,
Masanobu
Uchiyama
bc and
Zhenfeng
Xi
*ad
aMolecular Sciences (BNLMS), Key Laboratory of Bioorganic Chemistry and Molecular Engineering of Ministry of Education, College of Chemistry, Peking University, Beijing, 100871, China. E-mail: zfxi@pku.edu.cn; Fax: +86-10-62751708; Tel: +86-10-62759728
bGraduate School of Pharmaceutical Sciences, The University of Tokyo, 7-3-1 Hongo, Bunkyo-ku, Tokyo 113-0033, Japan
cAdvanced Science Institute (ASI), RIKEN, 2-1 Hirosawa, Wako, Saitama 351-0198, Japan
dState Key Laboratory of Organometallic Chemistry, SIOC, CAS, Shanghai, 200032, China
First published on 26th August 2011
Abstract
An efficient and specific cleavage of the endo-C(sp2)-Si bond of silole rings was developed. The stable silole ring was effectively opened in an unexpected regioselectivity by using an AcOH/ROH system, in which the AcOH behaved as a catalyst and the ROH as a nucleophile. Depending on the nature of the substituents at the 2- and 5-positions of the silole ring, the cooperative effect of the AcOH/ROH system exclusively cleaved one of the two endo-C-Si bonds to afford silylbutadiene derivatives.
Introduction
Carbon-silicon bonds are key structural features of organic molecules, and thus selective cleavage of C–Si bonds has become one of the central issues in recent organic synthesis.1 Several excellent methods have been developed for the cleavage of C–Si bonds, including the C(sp3)-Si bond2–4 and the C(sp2)-Si bond,5 by applying either the transition-metal catalyzed protocol or the stoichiometric reaction with strong bases or acids. As our continuous interest in activation and synthetic applications of C–Si bonds,4 we commenced an investigation into possible methods to cleave the endo-C(sp2)-Si bonds of siloles, aiming at challenging the following facts: (1) A silole ring of the general type contains two kinds of C–Si bonds, the endo-C(sp2)-Si bond and the exo-C(sp3)-Si bond. Furthermore, if the silole ring is unsymmetrically substituted, two different endo-C(sp2)-Si bonds will be available; However, research on the cleavage of such C–Si bonds has not been reported in the literature, except a single case of 1,1-dimethoxysilole.6 (2) No method has been reported for the efficient opening of the well-known silole rings,7 which are much more stable than the linear non-conjugated C–Si bonds.1,2 Here, we present a selective cleavage of the endo-C(sp2)-Si bond of silole using an AcOH/ROH catalysis system8,9 in which the AcOH behaved as a catalyst and the ROH as a nucleophile.This protocol lead to unexpected specific cleavage of the C(sp2)-Si bond of siloles substituted with different groups at the α and α′ positions (Scheme 1). A bifunctional mechanism of this AcOH/ROH system was theoretically elucidated as well.
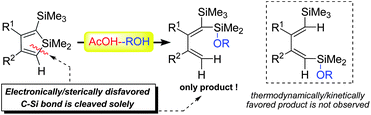 | ||
| Scheme 1 Specific cleavage of the endo-C(sp2)-Si bond of silole rings with AcOH/ROH bifunctional system. | ||
Results and discussion
In our initial attempts to open the silole ring of 1a, we found that those known methods used for the cleavage of C–Si bonds in the literature do not work well on siloles.1,4a–b,10 Surprisingly, however, we found that when a mixture of AcOH and CF3CH2OH was used, the silole ring could be opened via selective cleavage of one of the endo-C(sp2)-Si bonds. After careful evaluation of their ratios, we found that treating silole 1a with 15 mol% of AcOH in neat CF3CH2OH was the best reaction condition, affording the silylbutadiene 2aa as the sole product in 75% isolated yield (Entry 1, Table 1).
The pKa values of the components involved were also critical for this transformation. CCl3CH2OH, which has a similar pKa to CF3CH2OH, could generate the product 2ab, albeit in a lower yield (Entry 2), while alcohols with higher pKa values, such as EtOH (Entry 3), were ineffective. Similarly, the reaction proceeded with various phenols, including PhOH, p-MeO-PhOH, and p-CF3-PhOH, producing silylbutadienes 2ac–e in good yields (Entries 4–6). However, when phenols with stronger acidity, such as p-Ac- or p-NO2-PhOH, were used, no reaction took place. When acetate salt such as Cu(OAc)2 was used instead of AcOH, the same product 2a was obtained in similar yields.
Scheme 2 shows representative results of ring-opening reaction of symmetrically substituted siloles 1via cleavage of one of the two endo-C-Si bonds. All the ring-opening products, silylbutadienes 2b with all alkyl substituents, 2c with a fused ring, and phenyl-substituted 2d without substituents at the α and α′ positions of its butadienyl skeleton, were obtained in excellent yields from their corresponding siloles 1b–d. However, siloles 1e and 1f did not undergo ring-opening reactions even under severe reaction conditions, probably due to their chemically more stable conjugated structures and bulkiness.
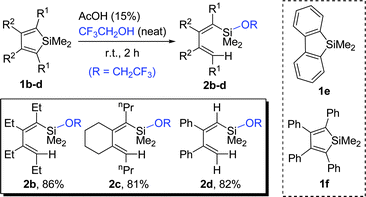 | ||
| Scheme 2 Ring-opening of symmetrically substituted silole derivatives 1 with CF3CH2OH. | ||
In the cases of siloles with different substituents at the α and α′ positions, the above cleavage of the endo-C(sp2)-Si bonds might raise regioselectivity issues. However, only one regioisomer was obtained for all such unsymmetrically substituted siloles, as illustrated in Scheme 3. This method was even successfully applied for the ring-opening of structurally more complicated silole 3e to afford the product 4e in 65% isolated yield, also as the only isomer.
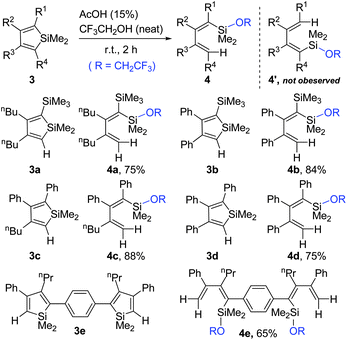 | ||
| Scheme 3 Ring-opening of unsymmetrically substituted siloles 3. | ||
The selectivity is in sharp contrast to the expected orientation according to the traditional manners for acid-catalyzed C(sp2)-Si bond cleavage,4a–b,10 as shown in Scheme 4. Taking 3a as an example, the path through intermediate 5a should be disfavoured while the one through 5a′ should be favored, if considering the following facts that 1) β-effect from the SiMe3 group in 5a′ is a potent stabilizer of the cationic 3-position; 2) the crowded gem-position bearing two silyl group visibly lowers the stability of 4a.
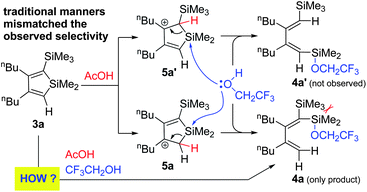 | ||
| Scheme 4 Mismatch between experimentally observed and theoretically expected products. | ||
DFT calculations at the b3lyp/6-31+g* level clearly support this view (Scheme 5).11 In the NBO analysis of intermediate 5A and 5A′, energy of the donor–acceptor interaction indicates high efficiency of the β-effect from the silyl group in stabilizing the neighbouring cation (Part I);12 this is probably the pivotal reason for the lower energy of 5A′ than that of 5A (Part II). Moreover, computed product analysis, i.e., comparison of the stability of the two possible products 4A and 4A′, shows the same trend (Part III). HOMO orbital analysis and charge calculation of silole also support the classical acid-catalyzed mechanism to afford 4′ (see ESI†). These results indicated that, thermodynamically and kinetically, the endo-Si-C(TMS) bond may be more reactive than the Si–C(H) one. Yet the experimental results are clearly opposite. This situation is somewhat similar to the fact that ipso-substitution of silyl arene through cleaving of C–Si bond was occasionally prevented by replacement of electrophilic substitutions in the para-position of silyl groups.13
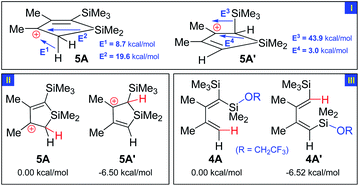 | ||
| Scheme 5 Second order perturbative estimates for donor–acceptor interaction by NBO analysis (Part I) and comparison of stability (Part II–III). (Energy data in Part I are stabilization energy from donor–acceptor interaction; energy data in Part II–III are total HF energy and those of 5A and 4A are set at zero, respectively). | ||
This led us to the idea of an acid–base cooperative mechanism of catalysis (Scheme 6).8,9 In this route, the acid–base complexation of AcOH with CF3CH2OH occurs at first.14a Several plausible structures for AcOH-CF3CH2OH complexes are computed to be energetically advantageous. Indeed, the 1H-NMR spectrum of a mixture of AcOH-CF3CH2OH (at the reaction concentrations) showed only a broad signal, and no signals corresponding to the OH group of AcOH or CF3CH2OH were observed, implying the existence of complexation and a very fast proton exchange equilibrium.14b
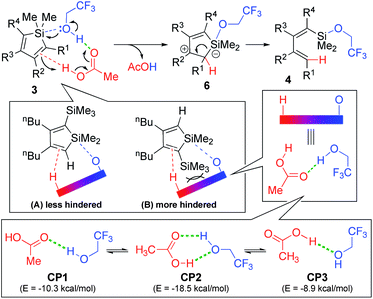 | ||
| Scheme 6 Proposed mechanism for the specific cleavage of the endo-C(sp2)-Si bond of siloles with the AcOH/ROH bifunctional system. (Energy of free AcOH and CF3CH2OH are set as zero). | ||
The AcOH-CF3CH2OH complex would then react with siloles, acting as an acid–base bifunctional system (Scheme 7). The access of the complex to the silole ring is assumed to be controlled mainly by steric effect. In cases where the α and α′ substituents are sterically highly unsymmetrical, such as 3a, the AcOH-CF3CH2OH complex plane would approach the silole ring perpendicularly to the side of less steric repulsion (A vs. B), which should lead to the observed regio-specificity. On the other hand, if the α and α′ positions were less sterically different, the direction of the ring opening would be electronically controlled, as the reaction of 3f, which gave 4f (with strong stabilization from SiMe3) instead of 4f′ (with weak stabilization from Me) as the sole product (Scheme 7).
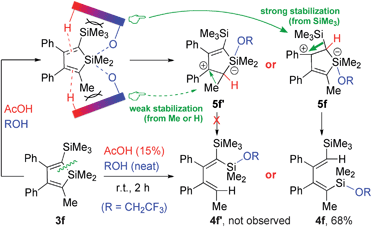 | ||
| Scheme 7 Electronically controlled ring-opening of silole 3f. | ||
Conclusions
In this work, we have developed the first method for the specific cleavage of the endo-C(sp2)-Si bond of silole ring under mild conditions, utilizing a simple combination of catalytic AcOH in CF3CH2OH. This reaction also provides a novel synthetic method for silylbutadiene-containing conjugated compounds, which are attractive candidates in many aspects.15,16 Further work to expand the scope of the reaction and to elucidate the reaction pathway with the help of theoretical and spectroscopic studies are in progress.Acknowledgements
This work was supported by the Natural Science Foundation of China and the Major State Basic Research Development Program (2011CB808705), and a JSPS Research Fellowship (to C.W.). Mr Fei Zhao did some experimental work. Helpful discussions with Prof. Xiaoqing Zhu of Nankai University and Prof. Ryo Takita in the University of Tokyo are appreciated. A generous allotment of computer time from RICC (RIKEN) is gratefully acknowledged.References
- For reviews, see: (a) G. R. Jones and Y. Landais, Tetrahedron, 1996, 52, 7599 CrossRef CAS; (b) A. K. Franz and K. A. Woerpel, Acc. Chem. Res., 2000, 33, 813 CrossRef CAS; (c) W. Rauf, A. Hazari, V. Gouverneur and J. M. Brown, Pure Appl. Chem., 2010, 82, 1415 CrossRef CAS; (d) T. J. Hiyama, J. Synth. Org. Chem. Jpn., 2010, 68, 729 CrossRef CAS.
- (a) A. Hosomi, Acc. Chem. Res., 1988, 21, 200 CrossRef CAS; (b) I. Fleming, J. Dunoguès and R. Smithers, Org. React., 1989, 37, 57 CAS; (c) I. Fleming, A. Barbero and D. Walter, Chem. Rev., 1997, 97, 2063 CrossRef CAS; (d) C. Mateo, C. Fernández-Rivas, A. M. Echavarren and D. J. Cárdenas, Organometallics, 1997, 16, 1997 CrossRef CAS; (e) W. S. Palmer and K. A. Woerpel, Organometallics, 1997, 16, 4824 CrossRef CAS; (f) A. K. Franz and K. A. Woerpel, J. Am. Chem. Soc., 1999, 121, 949 CrossRef CAS; (g) G. P. M. van Klink, H. J. R. de Boer, G. Schat, O. S. Akkerman, F. Bickelhaupt and A. L. Spek, Organometallics, 2002, 21, 2119 CrossRef CAS; (h) T. Matsuda, S. Kadowaki, Y. Yamaguchi and M. Murakami, Chem. Commun., 2008, 2744 RSC; (i) J. E. Cho, T. D. Senecal, T. Kinzel, Y. Zhang, D. A. Watson and S. L. Buchwald, Science, 2010, 328, 1679 CrossRef; (j) L. Chu and F.-L. Qing, Org. Lett., 2010, 12, 5060 CrossRef CAS; (k) L. Chu and F.-L. Qing, J. Am. Chem. Soc., 2010, 132, 7262 CrossRef CAS.
- (a) W. Rauf and J. M. Brown, Angew. Chem., Int. Ed., 2008, 47, 4228 CrossRef CAS; (b) M. Tobisu, M. Onoe, Y. Kita and N. Chatani, J. Am. Chem. Soc., 2009, 131, 7506 CrossRef CAS; (c) Y. Nakao, M. Takeda, T. Matsumoto and T. Hiyama, Angew. Chem., Int. Ed., 2010, 49, 4447 CrossRef CAS.
- For our recent work on C–Si bond cleavage, see: (a) Z. Xi, X. Liu, J. Lu, F. Bao, H. Fan, Z. Li and T. Takahashi, J. Org. Chem., 2004, 69, 8547 CrossRef CAS; (b) Z. Xi, Z. Song, G. Liu, X. Liu and T. Takahashi, J. Org. Chem., 2006, 71, 3154 CrossRef CAS; (c) C. Wang, Q. Luo, H. Sun, X. Guo and Z. Xi, J. Am. Chem. Soc., 2007, 129, 3094 CrossRef CAS; (d) Q. Luo, L. Gu, C. Wang, J. Liu, W.-X. Zhang and Z. Xi, Tetrahedron Lett., 2009, 50, 3213 CrossRef CAS; (e) Q. Luo, C. Wang, L. Gu, W.-X. Zhang and Z. Xi, Chem. Asian J., 2010, 5, 1120 CAS; (f) Y. Liang, S. Zhang and Z. Xi, J. Am. Chem. Soc., 2011, 133, 9204 CrossRef CAS.
- For reviews on transition-metal catalyzed cleavage of C(sp2)-Si bonds, see: (a) T. Hiyama, in Metal-Catalyzed Cross-Coupling Reactions; F. Diederich, P. J. Stang, ed.;Weinheim, Germany, 1998 Search PubMed; (b) T. Hiyama, J. Organomet. Chem., 2002, 653, 58 CrossRef CAS; (c) S. E. Denmark and R. F. Sweiss, Acc. Chem. Res., 2002, 35, 835 CrossRef CAS; (d) A. C. Spivey, C. J. G. Gripton and J. P. Hannah, Curr. Org. Synth., 2004, 1, 211 CrossRef CAS; (e) S. E. Denmmark and J. D. Baird, Chem.–Eur. J., 2006, 12, 4954 CrossRef. For representative examples, see: (f) Y. Nakao, H. Imanaka, A. K. Sahoo, A. Yada and T. Hiyama, J. Am. Chem. Soc., 2005, 127, 6952 CrossRef CAS; (g) E. Alacid and C. Nájera, Adv. Synth. Catal., 2006, 348, 2085 CrossRef CAS; (h) S. Yang, B. Li, X. Wan and Z. Shi, J. Am. Chem. Soc., 2007, 129, 6066 CrossRef CAS; (i) X. Dai, N. A. Strotman and G. C. Fu, J. Am. Chem. Soc., 2008, 130, 3302 CrossRef CAS; (j) L. Zhang and J. Wu, J. Am. Chem. Soc., 2008, 130, 12250 CrossRef CAS.
- When 1,1-dimethoxy-2,5-bis(trimethylsilyl)-3,4-diphenylsilole was treated with Br2, the ring-opened product, 1-bromo-4-bromodi(methoxy)silyl-1,4-bis(trimethylsilyl)-2,3-diphenyl-1,3-butadiene was obtained. See: J. Braddock-Wilking, Y. Zhang, J. Y. Corey and N. P. Rath, J. Organomet. Chem., 2008, 693, 1233 CrossRef CAS.
- For recent reviews on the synthesis and properties of siloles, see: (a) S. Yamaguchi and K. Tamao, J. Organomet. Chem., 2002, 653, 223 CrossRef CAS; (b) M. Hissler, P. W. Dyer and R. Reau, Coord. Chem. Rev., 2003, 244, 1 CrossRef CAS; (c) S. Yamaguchi, C. Xu and T. Okamoto, Pure Appl. Chem., 2006, 78, 721 CrossRef CAS; (d) R. West, Pure Appl. Chem., 2008, 80, 563 CrossRef CAS; (e) J. C. Sanchez and W. C. Trogler, Macromol. Chem. Phys., 2008, 290, 1527 CrossRef; (f) J. Chen and Y. Cao, Acc. Chem. Res., 2009, 42, 1709 CrossRef CAS; (g) J. Ohshita, Macromol. Chem. Phys., 2009, 210, 1360 CrossRef CAS; (h) M. Wang, G. Zhang, D. Zhang, D. Zhu and B.-Z. Tang, J. Mater. Chem., 2010, 20, 1858 RSC; (i) W.-Y. Wong and P. D. Harvey, Macromol. Rapid Commun., 2010, 31, 671 CrossRef CAS.
- For recent reviews, see: (a) S. Saito and H. Yamamoto, Acc. Chem. Res., 2004, 37, 570 CrossRef CAS; (b) H. Yamamoto and K. Futatsugi, Angew. Chem., Int. Ed., 2005, 44, 1924 CrossRef CAS; (c) M. S. Taylor and E. N. Jacobsen, Angew. Chem., Int. Ed., 2006, 45, 1520 CrossRef CAS; (d) T. Hashimoto and K. Maruoka, Chem. Rev., 2007, 107, 5656 CrossRef CAS; (e) T. Ooi and K. Maruoka, Angew. Chem., Int. Ed., 2007, 46, 4222 CrossRef CAS; (f) D. H. Paull, C. J. Abraham, M. T. Scerba, E. Alden-Danforth and T. Lectka, Acc. Chem. Res., 2008, 41, 655 CrossRef CAS; (g) K. Ding, Chem. Commun., 2008, 909 RSC; (h) M. Shibasaki, M. Kanai, S. Matsunaga and N. Kumagai, Acc. Chem. Res., 2009, 42, 1117 CrossRef CAS; (i) N. Kumagai and M. Shibasaki, Angew. Chem., Int. Ed., 2011, 50, 4760 CrossRef CAS.
- For representative examples, see: (a) L. Wilczek and J. Chojnowski, Macromolecules, 1981, 14, 9 CrossRef CAS; (b) L. Wilczek and J. Chojnowski, Makromol. Chem., 1983, 184, 77 CrossRef CAS; (c) T. Motomura, K. Inoue, K. Kobayashi and Y. Aoyama, Tetrahedron Lett., 1991, 32, 4757 CrossRef CAS; (d) C. D′Silva and D. A. Walker, J. Org. Chem., 1998, 63, 6715 CrossRef; (e) M. Cypryk and Y. Apeloig, Organometallics, 2002, 21, 216 CrossRef; (f) M. Cypryk, J. Phys. Chem. A, 2005, 109, 12020 CrossRef CAS; (g) N. Susperregui, D. Delcroix, B. Martin-Vaca, D. Bourissou and L. Maron, J. Org. Chem., 2010, 75, 6581 CrossRef CAS; (h) T. Marcelli, Angew. Chem., Int. Ed., 2010, 49, 6840 CrossRef CAS; (i) V. N. Wakchaure and B. List, Angew. Chem., Int. Ed., 2010, 49, 4136 CrossRef CAS; (j) Z. Zhang, Z. Wang, R. Zhang and K. Ding, Angew. Chem., Int. Ed., 2010, 49, 6746 CrossRef CAS; (k) S. Xu, Z. Wang, Y. Li, X. Zhang, H. Wang and K. Ding, Chem.–Eur. J., 2010, 16, 3021 CrossRef CAS; (l) T. Kano, T. Kumano, R. Sakamoto and K. Maruoka, Chem. Sci., 2010, 1, 499 RSC.
- For reviews and examples, see: (a) M. Asaoka, K. Shima and H. Takei, J. Chem. Soc., Chem. Commun., 1988, 430 RSC; (b) I. Fleming, J. Dunogues and R. Smithers, Org. React., 1989, 37, 57 CAS; (c) C. Chuit, R. J. P. Corriu, C. Reye and J. C. Young, Chem. Rev., 1993, 93, 1371 CrossRef CAS; (d) P. F. Hudrlik, A. M. Kassim, E. L. O. Agwaramgbo, K. A. Doonquah, R. R. Roberts and A. M. Hudrlik, Organometallics, 1993, 12, 2367 CrossRef CAS; (e) M. Ahmar, C. Duyck and I. Fleming, Pure Appl. Chem., 1994, 66, 2049 CrossRef CAS; (f) B. A. Keay, Chem. Soc. Rev., 1999, 28, 209 RSC.
- To save the calculation resource, the simplified methyl-substituted models, 5A, 5A′, 4A and 4A′, were used instead of n-butyl-substituted 5aet al. All calculations were carried out using Gaussian 03; (Revision E.01, M.J. Frisch, et al., Gaussian, Inc., Wallingford CT, 2004). The GRRM method based by employing Gaussian program was also used for optimization of structures, see: K. Ohno and S. Maeda, Chem. Phys. Lett., 2004, 384, 277. The NBO analysis was done by using NBO 5.G. with Gaussian 03, see: E.D. Glendening, J.K. Badenhoop, A.E. Reed, J.E. Carpenter, J.A. Bohmann, C.M. Morales and F. Weinhold, NBO 5.G. Theoretical Chemistry Institute, University of Wisconsin, Madison, WI, 2004; http://www.chem.wisc.edu/%E2%88%BCnbo5. Full citation and calculation details are given in the ESI†.
- In Scheme 5, although E2 and E4 represent the same type of hyperconjugative interaction with almost the same geometry, yet they are quite different in value. This is presumably due to the difference of structures of 5A and 5A′. The dihedral angle of the cation p-orbital and the corresponding endo-C-Si bond in 5A′ is about 63.4°, while that in 5A is 57.3°. Such less paralleling orientation may cause less hyperconjugative interaction.
- (a) R. A. Benkeser and R. B. Currie, J. Am. Chem. Soc., 1948, 70, 1780 CrossRef CAS; (b) H. Ishibashi, H. Sakashita and M. Ikeda, J. Chem. Soc., Perkin Trans. 1, 1992, 1953 RSC; (c) M. Herrlich, N. Hampel and H. Mayr, Org. Lett., 2001, 3, 1629 CrossRef CAS.
- (a) The AcOH-ROH complex could have many potential formulas. To minimize computational requirements, in this paper we chose the simplest one as a representative combination (AcOH/ROH = 1/1); (b) The 1H-NMR spectra of mixtures of AcOH and different phenols showed similar patterns (see ESI†).
- (a) F. Zhao, S. Zhang and Z. Xi, Chem. Commun., 2011, 47, 4348 RSC; (b) D. Li, Y. Cao, A. Shi and Z. Xi, Chem.–Asian J., 2011, 6, 392 CrossRef CAS; (c) D. Li, G. Liu, Q. Hu, C. Wang and Z. Xi, Org. Lett., 2007, 9, 5433 CrossRef CAS; (d) Z. Xi, Acc. Chem. Res., 2010, 43, 1342 CrossRef CAS. see also: (e) J. L. Paih, C. V.-L. Bray, S. Derien and P. H. Dixneuf, J. Am. Chem. Soc., 2010, 132, 7391 CrossRef; (f) K. M. Buchner and K. A. Woerpel, Organometallics, 2010, 29, 1661 CrossRef CAS.
- (a) T. Y. Luh and K. T. Wong, Synthesis, 1993, 4, 349 CrossRef; (b) J. D. Winkler, Chem. Rev., 1996, 96, 167 CrossRef CAS; (c) The Chemistry of Dienes and Polyenes (Ed.:Z. Rappoport), Wiley, Chichester, 1997, Vol. 1 and 2001, Vol. 2 Search PubMed; (d) H. Meier, Angew. Chem., Int. Ed., 2005, 44, 2482 CrossRef CAS; (e) M. Mori, Adv. Synth. Catal., 2007, 349, 121 CrossRef CAS; (f) M. E. Welker, Tetrahedron, 2008, 64, 11529 CrossRef CAS; (g) V. P. Ananikov, D. V. Hazipov and I. P. Beletskaya, Chem.–Asian J., 2011, 6, 306 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Materials including experimental procedures, NMR spectra of all new products and computation details. See DOI: 10.1039/c1sc00356a |
| This journal is © The Royal Society of Chemistry 2011 |