Nickel-catalyzed cross coupling of non-activated alkyl halides: a mechanistic perspective
Xile
Hu
*
Laboratory of Inorganic Synthesis and Catalysis, Institute of Chemical Sciences and Engineering, École Polytechnique Fédérale de Lausanne (EPFL), SB-ISIC-LSCI, BCH 3305, Lausanne, CH 1015, Switzerland. E-mail: xile.hu@epfl.ch; Fax: +41 21693 9305; Tel: +41 24693 9781
First published on 15th August 2011
Abstract
Cross coupling of non-activated alkyl halides is a potentially transformative methodology in organic synthesis. Herein we review the recent development of nickel-catalyzed coupling of non-activated alkyl halides. The current understanding of the mechanism of these coupling reactions is highlighted. As the mechanism is ligand-dependant, the perspective is organized according to the types of ligands employed in the catalysis.
 Xile Hu | Xile Hu was born in 1978 in Fujian, China. He received a B.S. degree from Peking University (June 2000) and a Ph.D. degree from the University of California, San Diego (December 2004), both in chemistry. His dissertation “Metal Complexes of Tripodal N-Heterocyclic Carbene Ligands: Synthesis, Structure, Bonding, and Reactivity” was guided by Prof. Karsten Meyer. He became a postdoctoral scholar in the group of Prof. Jonas C. Peters at the California Institute of Technology in February 2005. At Caltech, he worked on Co-catalyzed hydrogen evolution. In 2007, he was appointed as a tenure-track assistant professor of chemistry at the École Polytechnique Fédérale de Lausanne (EPFL) in Switzerland. His research interest is in the development of catalysts based on earth-abundant elements for applications in synthesis and in energy and sustainability. He received a starting grant from the European Research Council (ERC) in 2010 and was awarded the Werner Prize of the Swiss Chemical Society in 2011. |
1. Introduction
Heck, Negishi, and Suzuki received the 2010 Nobel Prize in chemistry for their pioneering contributions in palladium-catalyzed cross coupling reactions.1 The award is a testament to the tremendous impact cross coupling chemistry has made in the field of organic synthesis. Today, C–C cross coupling is one of the most powerful tools for the construction of carbon-based molecules and materials.2 However, not all coupling reactions are developed to the same degree. With regard to the electrophilic coupling partner, aryl, vinyl, and alkynyl halides and pseudo-halides can be readily coupled using Pd catalysis.3 Conversely, alkyl halides, especially those containing β-hydrogens, remain difficult substrates.4–8 Based on the general catalytic cycle for cross coupling reactions (Fig. 1), the two most often cited causes for the difficulty in coupling of non-activated alkyl halides are: (1) alkyl electrophiles are more electron rich compared to their aryl and vinyl counterparts, and therefore they have less tendency to undergo oxidative addition with a metal catalyst. (2) If oxidative addition of alkyl halides occurs, the resulting metal alkyl intermediates suffer from unproductive β-H elimination and thus cross coupling is not efficient. In addition, alkyl halides are prone to many side reactions such as base-promoted HX elimination (X = halide) and halide exchange reactions under coupling conditions. For these reasons, the development of cross coupling of non-activated alkyl halides falls behind those of aryl and vinyl electrophiles.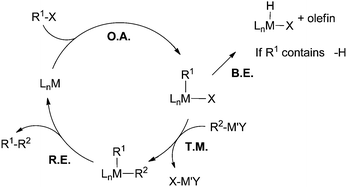 | ||
| Fig. 1 A general catalytic cycle for transition-metal-catalyzed C–C cross coupling reactions. O.A. = oxidative addition; B.E. = β-H elimination; T.M. = transmetalation; R.E. = reductive elimination. | ||
Kochi et al. studied the thermodynamics and kinetics of cross coupling reactions of alkyl halides in the early 1970s,9 and Suzuki et al. reported the first Suzuki-Miyaura coupling of non-activated alkyl halides in 1992.10 Yet general and preparative methods for cross coupling of alkyl halides started to emerge only in the mid to late 1990s.11 Significant progress has been made since then, resulting in many active catalytic systems. Several reviews, highlights, and accounts have been published on this topic.5–8,12–14 Fu et al. summarized the developments of Ni-catalysis up to 2004,6 and Beller et al. reviewed the state of the art of the field in 2005.7 Lautens et al. summarized coupling reactions of activated and non-activated secondary alkyl halides in 2009.8 These reviews were organized by the types of substrates and/or reactions. The mechanistic discussion was brief. Additionally, Cádenas et al. reviewed Ni-catalyzed Negishi coupling in 2009,14 which included the Negishi coupling of alkyl halides.
The majority of reported catalysts are based on Ni, Pd, Fe, Co, and Cu. Ni-based catalysts are the most versatile in terms of substrate scope and group tolerance. This perspective focuses on the latest developments in Ni-catalysis and the current understanding of the mechanism. It appears that there are multiple reaction pathways in Ni-catalyzed reactions. The mechanism of the catalysis depends on the specific ligands employed. Therefore, the review is organized according to the types of ligands. A brief introduction of the organometallic chemistry of nickel is also presented.
2. Organometallic chemistry of nickel
2.1 Oxidation states and coordination chemistry
The oxidation states of Ni span from 0 to +IV.15 Most Ni complexes are in the oxidation states 0, I, and II. Ni(0) is stabilized by soft and/or π-acceptor ligands such as CO, phosphine, N-heterocyclic carbene, bipyridene, and olefins. Ni(COD)2 (COD = 1,5-cyclooctadiene) and Ni(PPh3)4 are commonly used Ni(0) sources. Both compounds have a tetrahedral geometry. The COD and PPh3 ligands are labile and readily dissociate from Ni to form more reactive lower-coordinate species. 3- and 2-coordinate Ni(0) complexes have been isolated.Ni(I) complexes often contain phosphine or carbene ligands. They are mostly 4- or 5-coordinate complexes. Di- and polynuclear complexes with Ni(I) ions are also well-known. The Ni ion has a d9 configuration, and therefore mononuclear Ni(I) complexes are paramagnetic. Literature reports and our own experience show that it is possible to obtain well-resolved but paramagnetically shifted 1H NMR spectra of Ni(I) complexes. The assignment of the spectra is not trivial, but the spectra can be used as finger-prints to identify the various Ni(I) species involved. With S = 1/2, the electron paramagnetic resonance (EPR) spectra of Ni(I) complexes are relatively simple. The spectra are normally collected below room temperature (with liquid nitrogen or liquid helium) in order to have a higher resolution. Authentic Ni(I) species have an effective g value that is significantly deviated from 2.0 (e.g. 2.2), whereas an organic ligand radical typically has a g value close to 2.0. This difference can be used to distinguish a Ni(I) complex from a Ni(II)/ligand-radical complex.
Ni(II) complexes are ubiquitous. They can be 4, 5, or 6-coordinate. Square-planar Ni(II) complexes are diamagnetic; tetrahedral Ni(II) complexes are paramagnetic. 5 and 6-coordinate Ni(II) complexes are generally paramagnetic. With an integer spin (S = 1), paramagnetic Ni(II) complexes are hard to detect by conventional EPR spectroscopy. The identification and structural elucidation of these compounds rely on X-ray crystallography. UV-Vis spectroscopy and mass spectrometry may be used as well.
There are fewer Ni(III) and Ni(IV) complexes. K3NiF6 and M2NiF6 (M = alkali cation) are stabilized by electronegative F− ligands. Formal Ni(III) complexes with pincer ligands are known,16 and they are catalysts for Kharasch reactions. The oxidation state of Ni in these complexes is not yet thoroughly studied. Some macrocyclic Ni(III) and Ni(IV) complexes are structurally characterized, for example, complex 1 with a tetraanionic N4 ligand17 and complex 2 with two pyridinyl dioximato ligands (Scheme 1).18 A Ni(III) ion has a d7, t2g6eg1 electronic configuration. As such, it is subject to Jahn–Teller distortion. This is indeed found in K3NiF6 and [Ni(bipy)3]3+ (bipy = bipyridine), which show a tetragonal distortion.15
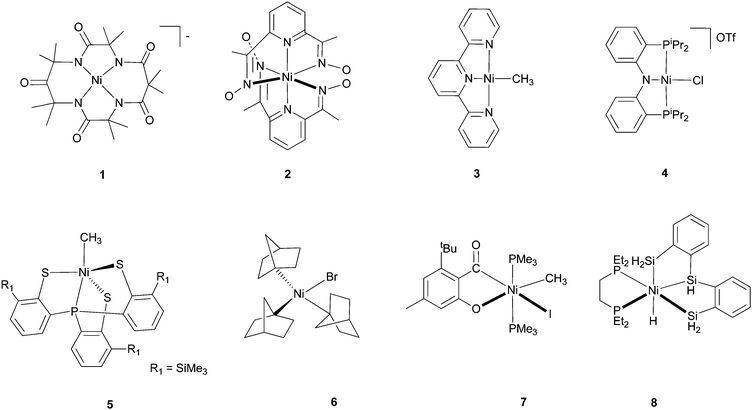 | ||
| Scheme 1 Representative Ni complexes. | ||
With redox active ligands such as pyridine, imine, aniline, thiolate, etc., the assignment of the oxidative states of Ni is not straightforward. [Ni(terpy)(CH3)] (3), which is formally a Ni(I) complex according to the simple charge balance consideration, is in fact a Ni(II) complex bound to a radical-anion ligand.19 Likewise, [Ni(PNP)Cl](OTf) (4), is a Ni(II) complex bound to a radical-cation ligand instead of a Ni(III) complex.20 Whether redox-active ligands facilitate Ni-mediated catalytic transformations is an open and interesting question. If the redox chemistry of ligands provides an easier access to otherwise too unstable intermediates, and if the ligand-based chemistry is not replaceable by metal-based redox chemistry, then one can imagine new principles for catalyst design using redox-active ligands.21 The “non-innocent” character of ligands in the redox chemistry of Ni complexes has long been revealed. For example, Busch et al. established in 1974 by EPR that diimine ligands were reduced in place of Ni(II) in a series of Ni complexes.22 Nevertheless, the involvement of ligand redox chemistry in an efficient Ni catalysis is less known. An excellent example was provided by Vicic et al.,19 who showed convincingly that terpyridine ligands are reduced to radical anions in Ni-catalyzed alkyl–alkyl Negishi coupling reactions (see section 4.5).
2.2 Organonickel chemistry
The most common organometallic Ni complexes are Ni(n) (n = 0–II) with CO, olefin, carbene ligands, and Ni(II) with alkyl, allyl, aryl, and hydride ligands. The latter complexes are normally prepared by transmetalation of Ni-halide complexes with a lithium or Grignard reagent, or a hydride source (e.g., LiBEt3H, LiAlH4). They can also be synthesized by oxidative addition of R–X or R–H on Ni(0) complexes. There are many Ni(II) π-ally complexes, e.g., [Ni(η3-C3H5)2]. These allyl complexes are nucleophilic, in contrast to Pd(II) π-ally complexes.Ni(I) and Ni(III) hydride and hydrocarbyl complexes are less often encountered. They are however of significant interest to the bio-organometallic community because such species are invoked as possible intermediates in the catalytic cycles of [NiFe]-hydrogenase, carbon monoxide dehydrogenase (CODH)/acetyl-CoA-synthetase (ACS), and methyl-coenzyme M reductase (MCR).23 Fascinatingly, a σ-alkane-Ni complex was identified as a catalytic intermediate for MCR.24 [Ni(CH3)(PS3)]− (5) is described as a genuine Ni(III) alkyl complex because the complex has a g value of 2.13.25 Organo-Ni(I) and Ni(III) species are also possible intermediates in cross coupling reactions.
Ni(IV) organometallic complexes are rare but not unprecedented. Complex 6 is distorted tetrahedral and is stable in the crystalline form for several days in air but decomposes rapidly in solution.26 Complex 7 is octahedral; it was proposed as an authentic Ni(IV) based on its NMR and structural features.27 The isolation of this complex is significant because it can serve as a mechanistic probe for the involvement of high-valent Ni species in homogeneous catalysis. The possibility that the acyl phenolate ligand is redox-active cannot be excluded, and might be worthy of re-investigation. The Ni(IV)–H complex 8 was said to exist in solution at low temperature; in the solid state, it existed as an η2-(Si–H)Ni(II) complex.28
The typical organometallic reactions such as oxidative addition, reductive elimination, migratory insertion, β-elimination, and transmetalation are well documented for Ni complexes. Oxidative addition of aryl, vinyl, and allyl halides and pseudo-halides is straightforward and is facilitated by prior coordination of Ni to the double bonds. Whereas oxidative addition of aryl halides to Pd(0) occurs by concerted three-centered transition states, Kochi et al. showed that analogous reactions to Ni(0) occurred by single-electron transfer from Ni(0) to aryl halide to form Ni(I) intermediates.29 Oxidative addition of a π–bond to Ni(0) to form two σ–bonds is also facile, and is applied to develop cyclization and reductive coupling reactions. Oxidative addition to Ni(II) is possible. For example, complex 7 is produced by oxidative addition of CH3I to a Ni(II) complex.27
Reductive elimination of four-coordinate Ni(II) complexes is accelerated by the coordination of an electron-deficient fifth ligand. Yamamoto et al.30 showed that Ni(alkyl)2(bipy) complexes are thermally stable. In the presence of electron-poor olefins (e.g.acrylnitrile, maleic anhydride), reductive elimination occurs to form alkyl–alkyl and Ni(bipy)(alkene)n quantitatively. The acceleration originates from the changes in the transition state energies and reactive intermediates. Reductive elimination from a trigonal bipyramidal complex leads to the occupation of a molecular orbital that is non-bonding; on the contrary, reductive elimination from a square-planar complex leads to occupation of a molecular orbital that is metal–ligand anti-bonding.31 This phenomenon can be exploited in cross coupling reactions. The first preparative method for alkyl–alkyl Negishi coupling took advantage of substrates containing remote double bonds, which presumably coordinated to Ni and promoted reductive elimination (section 3.1).11
3. Catalysis employing olefin ligands
3.1 Olefins as ligands
 | ||
| Scheme 2 Ni-catalyzed alkyl–alkyl Negishi coupling of substrates with pendant double bonds. | ||
Knochel et al. hypothesized that the double bonds in these substrates coordinated to Ni to promote alkyl–alkyl reductive elimination.32 If this is the case, then an external unsaturated molecule may function as a promoter as well. Indeed, his group soon developed a method to couple alkyl iodides without a pendant double or triple bond to dialkylzinc reagents (Scheme 3).33 π-acceptor ligands such as acetophenone, benzophenone, or styrene derivatives were used as additives, and 3-trifluoromethylstyrene was found to be the most efficient. A large number of functional groups were tolerated. Similar methods were subsequently developed for the coupling of primary alkyl iodides with arylzinc bromides,34 benzylzinc bromides,35 and alkylzinc halides.36 For the coupling of benzylzinc and alkylzinc bromides, Bu4NI was required as an additive. It was suggested that Bu4NI either increased the ionic strength of the medium, or formed more reactive ‘ate’ complexes with organozinc halides.
 | ||
| Scheme 3 Ni-catalyzed Negishi coupling using 3-trifluoromethylstyrene as an additive. | ||
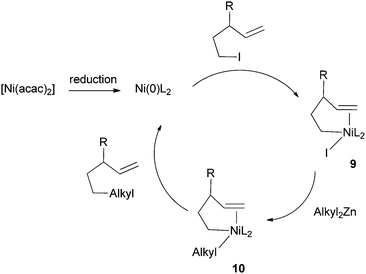 | ||
| Scheme 4 Mechanism of olefin-assisted Ni-catalyzed Negishi coupling. | ||
3.2 Dienes as ligands
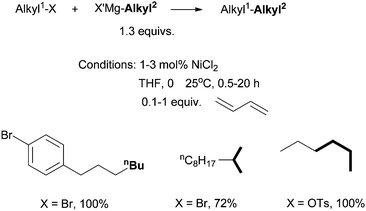 | ||
| Scheme 5 Ni-catalyzed Kumada coupling of alkyl halides using diene ligands. | ||
For analogous coupling of alkyl halides with organozinc reagents, a tetraene additive (9 mol%) was required.43 Only diorganozinc reagents could be used, and 3 equiv. of MgBr2 was needed to activate the zinc reagents.
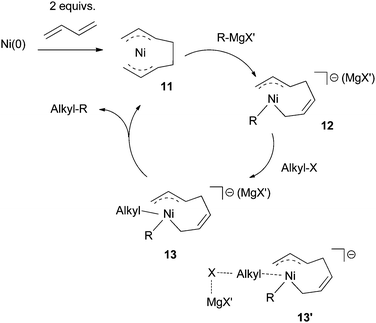 | ||
| Scheme 6 Proposed mechanism for Ni-catalyzed Kumada coupling using diene ligands. | ||
The Ni(0)/Ni(II) cycle was ruled out by Kambe et al. because Ni(COD)2 did not react with octyl bromide in the presence of 1,3-butadiene.37 The intermediacy of nickel-π-allyl species was supported by the fact that Ni(allyl)2 and Pd(allyl)2 complexes were competent catalysts under similar conditions.39 The Pd(allyl)2 complex reacted with Et–MgCl at −60 °C in THF to form a Pd(alkyl)(η1-allyl)(η3-allyl) species (14), which then reacted with hexyl bromide to form octane in a 38% yield (Scheme 7).39 Analogous reactions on the Ni(allyl)2 system would have given more insights in the mechanism of the Ni-catalysis, however, these reactions were not reported.
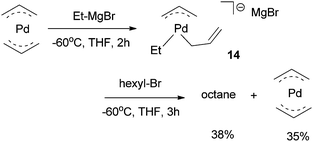 | ||
| Scheme 7 Reactivity of Pd(allyl)2 complex. | ||
The coupling of (bromoethyl)cyclopropane with n-octylBr using this protocol produced nonylcyclopropane in an 87% yield (Scheme 8). 1-Dodecene, which might arise from the ring opening of the cyclopropylmethyl radical, was not detected.37 This result rules out a radical mechanism for the activation of alkyl halides. An SN2-type mechanism is more likely. Kambe et al. also developed Cu-catalyzed coupling of alkyl halides with aryl Grignard reagents.44 The reaction of α,β-d2-β-adamantylethyl chloride occurred primarily with an inversion of configuration with a 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 selectivity (Scheme 9). This result is consistent with an SN2-mechanism in the Cu-catalysis. The same study was not carried out for the Ni-catalysis.
:1 selectivity (Scheme 9). This result is consistent with an SN2-mechanism in the Cu-catalysis. The same study was not carried out for the Ni-catalysis.
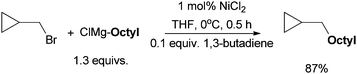 | ||
| Scheme 8 Coupling reaction using a radical-probe substrate. | ||
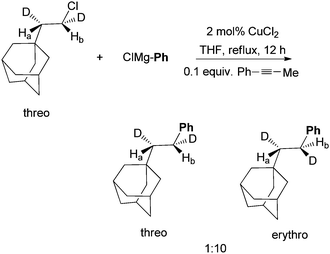 | ||
| Scheme 9 Probe of stereochemistry in Cu-catalyzed coupling. | ||
Compared to Ni- or Pd-catalyzed cross coupling of aryl halides, the mechanism in Scheme 6 has two distinguishing features: (1) transmetalation occurs before the oxidative addition of electrophiles; (2) a Ni(IV) intermediate is invoked.
A DFT study (B3LYP/DZVP, implicit solvation: SCRF = PCM, THF, 243 K) was conducted on the coupling of EtBr with EtMgCl using Ni(allyl)2 as the catalyst.45 The study only considered the mechanism in Scheme 6, but not possible alternatives. It was proposed that the Mg cation had a significant interaction with the allyl ligands, and this interaction was important for the catalysis. Oxidative addition was calculated as the rate-determining step. The calculated reaction barrier of 138 kJ mol−1, however, was far from the experimental value, as the coupling occurred at −30 °C.
4. Catalysis employing pyridine and imine ligands
4.1 Pybox as ligands
Fu et al. reported in 2003 the first Ni-catalyzed Negishi coupling of secondary alkyl halides with alkylzinc reagents (Scheme 10).46 Only Pybox-based (Pybox = pyridine bisoxazoline) ligands were effective. Phosphines and carbenes failed to generate any product. The s-butyl-derivative of Pybox provided higher yields than the i-propyl- and t-butyl-derivatives. The reaction conditions were compatible to many functional groups such as sulfonamide, ester, amide, etc.NiBr2 gave lower yields than Ni(COD)2. Pd(0) and Pd(II) complexes gave no coupling products. Fu et al. speculated that the chelating nature of Pybox ligand made β-H elimination unfavorable due to coordinative saturation. This hypothesis remains to be directly confirmed using an isolated Ni-Pybox alkyl complex. | ||
| Scheme 10 Ni-catalyzed Negishi coupling of secondary alkyl halides using a Pybox ligand. | ||
Fu et al. subsequently developed Ni-catalyzed enantioselective coupling of racemic and activated secondary alkyl halides with alkylzinc reagents using Pybox ligands. For the coupling of α-bromoamides,47NiCl2/(R)-iPr-Pybox was best; for the coupling of benzylic bromides,48 NiBr2·diglyme/(S)-iPr-Pybox was best; for the coupling of allylic chlorides,49 NiCl2·glyme/(s)-BnCH2-Pybox was best; and for coupling of α-bromoketones,50 NiCl2·glyme/(+)-MeOCH2-Pybox and bis(oxazoline) was best. Fu et al. also reported asymmetric coupling of propargylic halide with arylzinc reagents using Ni/Pybox catalysis.51 Gagné et al. applied the Ni/Pybox-catalysis for the synthesis of alkylated glycosides.52 Caeiro et al. reported asymmetric coupling of benzylic bromides with trialkynylindium reagents using NiBr2·diglyme/(S)-iPr-Pybox.53 Leigh et al. developed a Ni-Pybox-catalyzed homocoupling of alkyl bromides. They applied the method for a template synthesis of rotaxanes.54
Cádenas et al. employed the Ni/Pybox method for the cascade formation of C(sp3)–C(sp3) bonds (Scheme 11).55Alkyl iodides containing an alkene group first underwent intra-molecular ring-closing reactions before being coupled to alkylzinc halides.
 | ||
| Scheme 11 Cascade formation of C–C bonds using Ni-Pybox catalysis. | ||
In an interesting development, Gong et al. reported Ni-catalyzed reductive cross coupling of two different alkyl halides using Pybox ligands (Scheme 12).56 The reactions required 3 equiv. of one coupling partner, and 3 equiv. of Zn. Stoichiometric reactions showed that alkyl bromides were not converted into alkylzinc reagents under the coupling conditions, although alkyl iodides might be converted into the zinc reagents. This protocol avoids the use of organometallic reagents, and exhibits a high group tolerance, for instance, towards alcohol and keto groups. The main drawback is the requirement for an excess of one coupling partner. The problem is less severe if this coupling partner is cheap, abundant, and can be easily separated from the product.
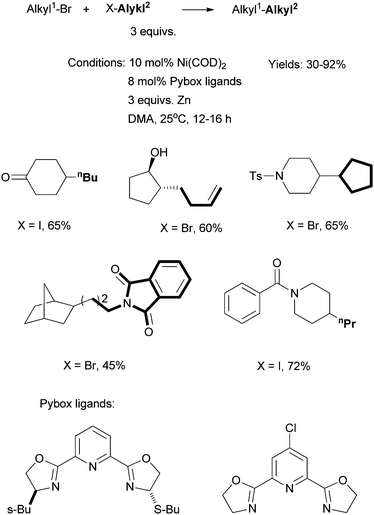 | ||
| Scheme 12 Ni-catalyzed reductive coupling of alkyl halides using a Pybox ligand. | ||
4.2 Phen as ligands
Fu et al. described in 2004 the first efficient method for Suzuki coupling of non-activated secondary alkyl halides with aryl boronic acids (Scheme 13).57 The combination of Ni(COD)2 precatalyst and bathophenanthroline ligand was essential. The Ni/Pybox system was not ineffective. Fu et al. then developed a Ni/phen system for the Hiyama coupling of secondary alkyl bromides with trifluorosilanes.58 An excess of CsF (4 equiv.) was required. 1,10-Phenanthroline and 2,2-bipyridine ligands gave slightly lower yields.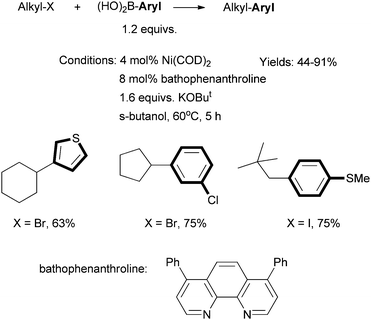 | ||
| Scheme 13 Ni-catalyzed Suzuki coupling of secondary alkyl halides using a phen-type ligand. | ||
4.3 Bipy as ligands
Fu et al. reported in 2005 a Ni-catalyzed cross coupling non-activated alkyl halides with trichloroaryltin reagents (Scheme 14).592,2′-Bipyridine was the best ligand; bathophenanthroline gave lower yields. The reactions required 10 mol% NiCl2, 15 mol% bipy, 7.0 equiv. of KOBut, and heating at 60 °C for 12 h.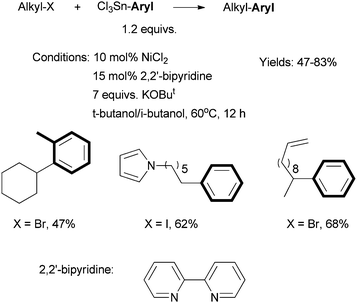 | ||
| Scheme 14 Ni-catalyzed Stille coupling of secondary alkyl halides using a bipy ligand. | ||
Weix reported Ni-catalyzed reductive cross coupling of aryl halides with alkyl halides (Scheme 15).60 A combination of a bipyridine ligand, a bis(phosphine) ligand, and pyridine gave optimized yields. 2 equiv. of Mn(0) was used as the reductant, and the in situ formation of an organo-Mn intermediate was excluded. The reactions were more selective if at least one of the coupling partners was an organic iodide. Alcohol, ester, amide, and keto groups were tolerated. The advantage of this coupling method is to avoid the need for preformed organometallic reagents.
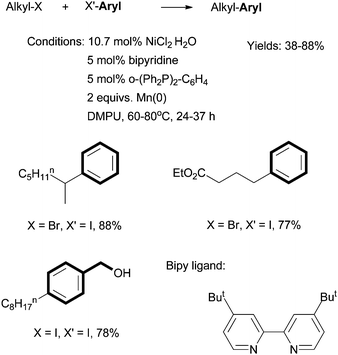 | ||
| Scheme 15 Ni-catalyzed reductive coupling of alkyl halides using a bipy ligand. | ||
4.4 Terpy as ligands
Vicic et al. reported in 2004 the alkyl–alkyl Negishi coupling catalyzed by [Ni(terpy)(CH3)] (terpy = terpyridine) (Scheme 16).61 The mechanistic implications of this system are particularly interesting (see section 4.5).19,62 The [Ni(terpy)(CH3)] complex was the first isolated and structurally characterized Ni catalyst for alkyl–alkyl Negishi coupling.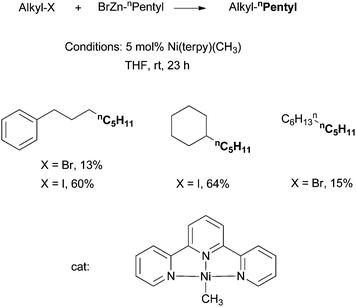 | ||
| Scheme 16 Ni-catalyzed alkyl–alkyl Negishi coupling using a Ni-terpy complex. | ||
Fu et al. developed in 2008 a NiCl2·glyme/terpy system for the cross coupling of secondary propargylic halides with secondary alkyl zinc reagents.63 Weix et al. developed in 2010 a Ni-catalyzed reductive dimerization of alkyl halides, tosylates, trifluoroacetates using a terpy ligand.64 The reactions could be run under air as Mn(0) was used as the reductant.
4.5 Mechanism
As Pybox, phen, bipy, and terpy ligands have similar structural and electronic properties, the mechanism of Ni-catalyzed cross coupling reactions employing these ligands should be similar to one another. Many aspects of the mechanism have been revealed and they are presented in the following paragraphs.First of all, plenty of evidence shows that the activation of alkyl halides occurs via a radical mechanism. Fu et al. reported that coupling of both exo- and endo-2-bromonorbornane with phenyboronic acid gave predominantly the exo-product (Scheme 17),57 suggesting that the same planar carbon radical intermediate was involved. The same results were obtained on the Ni-catalyzed Hiyama coupling.58 Fu et al. also reported the Stille coupling of substrates 15 (Scheme 18).59 The formation of the cyclized product is consistent with the initial formation of a secondary alkyl radical which cyclizes before reacting with Ni. The cyclization/coupling cascade was exploited further by Cádenas et al. (Scheme 12).55 The enantioselective cross coupling developed by Fuet al. proceeds by stereo-convergence of racemic starting materials rather than by kinetic resolution. The success of these asymmetric reactions suggests that both enantiomers of the secondary alkyl halides react to give the same planar carbon radical intermediates.
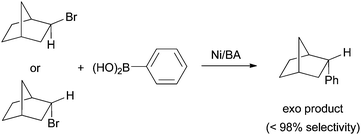 | ||
| Scheme 17 Exo -selectivity in Ni-catalyzed coupling. | ||
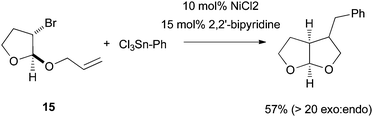 | ||
| Scheme 18 Ring-close rearrangement in Ni-catalysis. | ||
Vicic et al. examined the minor products for the reactions in Scheme 16.61 They found Ph(CH2)3-(CH2)3Ph and dicyclohexyl from the reactions of Ph(CH2)3-I and cyclohexyl-I, respectively. These molecules most likely originated from the dimerization of the corresponding alkyl radicals. When a radical clock, iodomethylcyclopropane was used as the substrate, olefinic products were observed, indicating the formation of cyclopropylmethyl radical which underwent a fast ring-open reaction. Cádenas et al. examined a number of radical-clock substrates in the Ni/Pybox system,55 and found that the combination of the alkyl radical with the Ni center had a rate that was comparable to a uni-molecular reaction with a rate of about 107 s−1. Cádenas et al. also found that the stable radical TEMPO inhibited the catalysis, again indicating that the oxidative addition of alkyl halides occurred via a radical intermediate.
Vicic et al. synthesized and isolated several Ni-terpy complexes and used them to probe the active species in the Ni-catalysis.62 The Ni(terpy)(CH3) complex (16 and 17) was prepared by reaction of [TMEDANi(CH3)2] (TMEDA = N,N,N′,N′-tetramethylethylenediamine) with terpyridines (Scheme 19). The by-product was a half equivalent of ethane. Reaction of Ni(COD)2 with a terpy ligand in the presence of CH3I gave the cationic [Ni(terpy′)(CH3)]I (18). 16–18 are all competent pre-catalysts for alkyl–alkyl Negishi coupling, with yields of 60–80% for alkyl iodides. Reaction of 18 with 5 equiv. of heptylZnBr gave the coupling product octane in only an 8% yield. This result suggests that 18, or similar Ni(II) species, is not the active species for the catalysis. Reaction of 17 with 5 equiv. of heptyl-I gave octane in a 90% yield, similar to that of the catalytic reaction. Thus, 17 and analogous neutral complexes having the formula of NiL(Alkyl) are active intermediates in Ni-catalyzed alkyl–alkyl coupling.
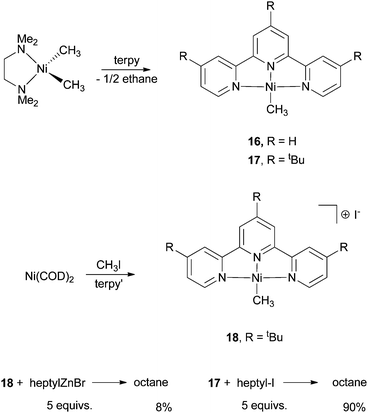 | ||
| Scheme 19 Reactivity of Ni-terpy complexes. | ||
Vicic et al. studied the electronic structure of complex 16.19 They found that this complex is best described as a Ni(II) complex of a reduced terpyridinyl ligand anion rather than a Ni(I) complex. Even though 16 and related complexes have been widely referred to as Ni(I) complexes in the literature, based on the work of Vicic et al., we suggest these complexes be called Ni(II)-ligand anion complexes in relevant mechanistic discussions. The particular electronic structure of these complexes may have a bearing on their catalytic efficiency. Because of the similarity between the terpyridynyl ligand with other imine- and pyridine-based ligands, similar Ni(II)-ligand anion complexes should be the active intermediates in reactions employing the latter ligands.
An overall catalytic cycle can be constructed for the reactions outlined in this section (Scheme 20). Terpyridine is used as the illustrative ligand here. Starting from the neutral alkyl complex [Ni(II)(L−)(Alkyl2)] (19), activation of alkyl halide takes place via single-electron-transfer to form [Ni(II)(L)(Alkyl2)]X (20) and an alkyl radical. Recombination of the carbon radical with Ni gives [Ni(L)(Alkyl1)(Alkyl2)]X (21), presumably a Ni(III) complex. Reductive elimination then provides the coupling product and [Ni(L)X] (22). Vicic et al. found that this Ni-halide complex is indeed Ni(I). Transmetalation of 22 with the alkyl nucleophile regenerates 19, with a concomitant intra-molecular electron transfer from Ni to the terpyridine ligand.

Phillips et al. used DFT (B3LYP/6-31G*+LANL2DZ; implicit solvation: PCM, THF) to reproduce the catalytic cycle in Scheme 20.65 According to their computations, this mechanism is feasible. The calculations focused on the reaction energy and activation barriers, but not on the electronic structures of the catalytic intermediates. The [Ni(terpy)(CH3)] complex was incorrectly referred to as Ni(I). A major disagreement between computation and experiment is the geometry of [Ni(II)(terpy)(CH3)]I. Vicic et al. showed that it is square-planar and cationic.61,62 The computation showed it was five-coordinate. The coordination geometry has a significant impact in the energy and property of the Ni(II) complex. A square planar complex is diamagnetic, and a 5-coordinate complex is paramagnetic (triplet). Phillips et al. did not discuss which and how spin-states of Ni complexes were treated in their computations.
5. Catalysis employing amino alcohol and diamine ligands
5.1 Amino alcohols as ligands
Fu et al. reported in 2006 the use of readily available amino alcohols as ligands for the Suzuki coupling of non-activated secondary alkyl halides with arylboronic acids (Scheme 21).66Prolinol permits the coupling of secondary alkyl chloride for the first time. The reactions were shown to go by a radical mechanism. Fu et al. then reported in 2007 the Hiyama coupling of activated and non-activated secondary alkyl halides with trifluoroarylsilanes using the Ni/norephedrine system (Scheme 22).67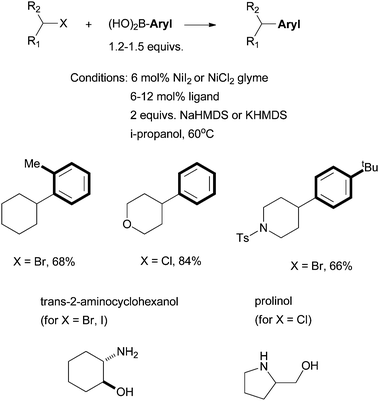 | ||
| Scheme 21 Ni-catalyzed Suzuki coupling of secondary alkyl halides using amino alcohol ligands. | ||
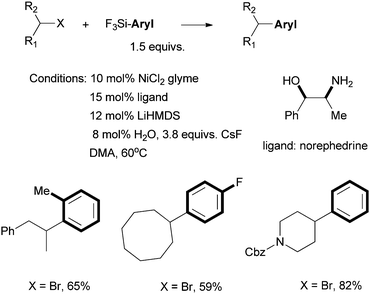 | ||
| Scheme 22 Ni-catalyzed Hiyama coupling of secondary alkyl halides using norephedrine as a ligand. | ||
5.2 Diamines as ligands
Fu et al. developed in 2007 a Ni-catalyzed Suzuki coupling of non-activated secondary alkyl bromides and iodides with alkyl boranes using a diamine ligand (Scheme 23).68 KOBut and tBuOH were shown to activate alkyl boranes for transmetalation with nickel. The trans-N,N′-dimethyl-1,2-cyclohexanediamine was the best ligand; its cis-isomer gave diminished yields (ca. 40%). Non-methylated 1,2-cyclohexanediamine and tetra-methylated trans-N,N,N′,N′-tetramethyl-1,2-cyclohexanediamine were not effective ligands. These results suggest that a secondary amine ligand is important for the efficiency of the catalysis. Fu et al. extended the system to the coupling of non-activated secondary alkyl chlorides using (R,R)-N,N′-dimethyl-1,2-diphenyl-1,2-ethylenediamine as the ligand.69 A similar system was applied for the asymmetric coupling of racemic α-bromoesters with trimethoxyarylsilanes.70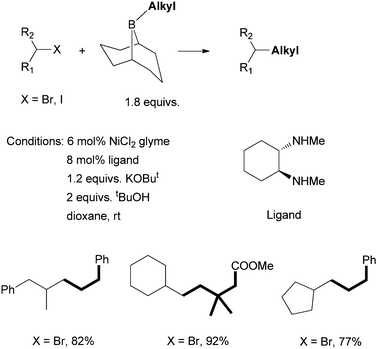 | ||
| Scheme 23 Ni-catalyzed Suzuki coupling of secondary alkyl halides using a diamine ligand. | ||
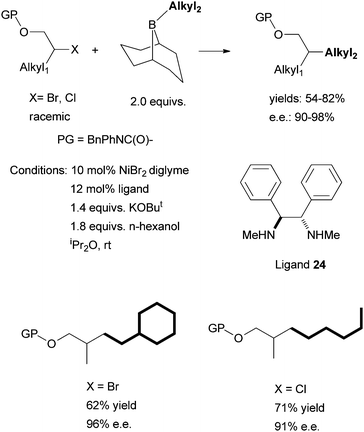
The use of chiral diamine ligands opens the door to the asymmetric cross coupling of non-activated alkyl halides. Indeed, in 2008, Fu et al. reported the first method for this type of coupling. Non-activated homobenzylic bromides were coupled to alkyl boranes (Scheme 24).71 The particular diamine ligand 23 was found to give the best enantioselectivity; similar diaryl-substituted diamine ligands could be used, but gave lower ee. An electron-donating group on the aryl ring of the homobenzylic halides led to higher ee than an electron-withdrawing group. Fu et al. hypothesized that the secondary interaction between the Ni catalyst and the CH2Ar substituent of the substrate was the origin of the high enantioselectivity. Changing CH2Ph to CH2CH2Ph decreased the ee significantly (from 90% to 14%).
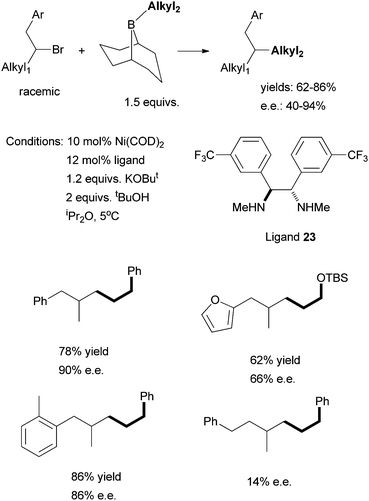
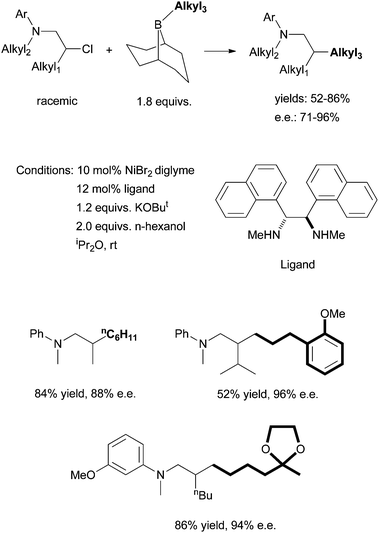
Following this development, Fu et al. reported in 2010 the asymmetric Suzuki coupling of racemic acylated halohydrins with alkyl boranes (Scheme 25).72 A similar Ni/diamine system was employed as the catalyst. The best ligand was 24. The interaction of the ether oxygen with the nickel catalyst was said to be important for the high selectivity. And then in 2011, Fu et al. reported the asymmetric Suzuki coupling of racemic secondary alkyl chlorides bearing proximal arylamines (Scheme 26).73 A structure-selectivity study indicated that the primary site of coordination of the arylamines is the nitrogen rather than the aromatic ring. Kinetic data suggested a rate-determining transmetalation step.
 | ||
| Scheme 26 Ni-catalyzed asymmetric coupling of secondary alkyl halides bearing pendant nitrogen atoms. | ||
6. Catalysis employing a pincer bis(amino)amide ligand
6.1 The pincer N2N ligand
Our group developed a new pincer N2N ligand Lockamine (Scheme 27).74–76 The ligand is based on a bis(aryl)amido framework to enforce high stability and structural rigidity. The amine donors were chosen to better match the ionic character of first-row transition metals. Nevertheless, the ligand forms stable complexes with first-to-third row transition metals such as Ni, Pd, Pt, and Ru.77 In these complexes, the Lockamine ligand adopts the expected meridional geometry. The complex bis[(2-dimethylamino)phenyl]amine nickel(II) chloride (Nickamine, 25, Scheme 27),78 underwent salt metathesis reactions to form a series of nickel complexes bearing alkyl, aryl, alkoxide, thiolate, and triflate ligands.75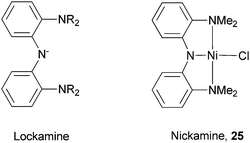 | ||
| Scheme 27 Lockamine and Nickamine ligands. | ||
6.2 Alkyl-alkyl Kumada coupling using Nickamine as catalyst
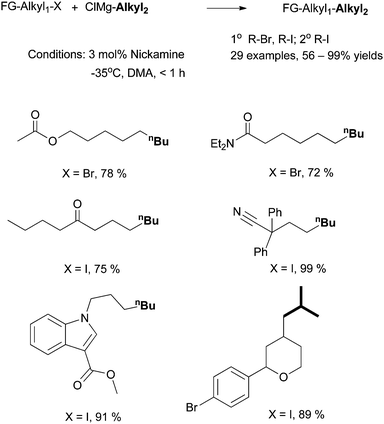 | ||
| Scheme 28 Alkyl-alkyl Kumada coupling by Nickamine. | ||
Aryl halides could not be coupled under the same experimental conditions. Thus, the oxidative addition of alkyl halides did not occur through a concerted process. Alkyl triflates and tosylates could not be coupled, which suggests that an SN2-type mechanism is unlikely. In fact, the result from the coupling of a radical-probe substrate indicates that the activation of alkyl halides takes place through a radical mechanism (Scheme 29).75 When bromomethylcyclopropane was coupled to n-pentylMgCl, 1-nonene was formed as the major product. The compound was presumably produced from the coupling of a 3-butenyl radical which was formed by the ring-opening isomerization of a cyclopropylmethyl radical.
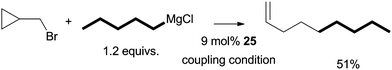 | ||
| Scheme 29 Coupling using a radical-probe substrate. | ||
The currently favored catalytic cycle is shown in Scheme 30. The nickel halide complex (26) is alkylated by a Grignard reagent to form a Ni(II) alkyl complex (27). The halide of the substrate first approaches 27 to mediate a single-electron transfer (SET). A one-electron oxidized complex (28) and an alkyl radical are produced after the electron transfer. The alkyl radical recombines with the nickel center to form the bis(alkyl) complex (29) which is probably six-coordinate. Reductive elimination then gives the coupling product and regenerates the nickel halide complex. The oxidation states of 28 and 29 can be ambiguous. 28 could be a Ni(III) complex, or a Ni(II) complex with an oxidized ligand. There is a good possibility that the N2N ligand can be readily oxidized, as Mindiola et al. showed that the analogous PNP ligand in [Ni(PNP)Cl](OTf) (4) is a radical-cation.20 Likewise, the oxidation state of Ni in 29 can be +2, +3, or +4 depending on the oxidation state of the ligand. Isolation and characterization of complexes resembling 28 and 29 will be of significant interest.
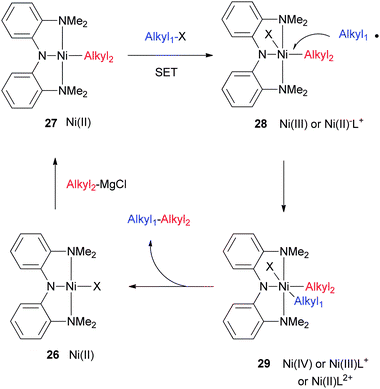
This mechanism is supported by the following experimental observations: (1) both Nickamine and [(MeN2N)Ni(CH3)] complexes are catalytically competent, confirming the roles of intermediates like 26 and 27. (2) Stoichiometric reactions are consistent with the mechanism. The reaction of 26 with an alkyl Grignard generates cleanly a Ni(II) alkyl complex which can be isolated and characterized. The reaction of a nickel alkyl complex with an alkyl halide generates the coupling product and a nickel halide complex in nearly quantitative yields. (3) The catalysts can be isolated after the reactions.79 If an excess of alkyl Grignard reagent is used, then at the end of the catalysis, a nickel alkyl complex can be observed as the only metal-containing species. If an excess of an alkyl iodide is used, then at the end of the catalysis, [(MeN2N)Ni(Cl)] (25) is observed as the only metal-containing species. According to the mechanism shown in Scheme 30, one expects to isolate [(MeN2N)Ni(I)] instead. A separate experiment showed that [(MeN2N)Ni(I)] reacts quickly with MgCl2 to form [(MeN2N)Ni(Cl)] under similar conditions.79 This reaction explains why [(MeN2N)Ni(Cl)] is observed in the end.
The efficiency of Nickamine in this alkyl–alkyl Kumada coupling indicates that the relevant Ni-alkyl complexes do not suffer from β-H elimination. One can imagine two origins for the lack of β-H elimination: (1) the square-planar [(MeN2N)Ni(alkyl)] complex is saturated and does not provide an open coordination site for β-H elimination. (2) β-H elimination is kinetically accessible but thermodynamically uphill. We studied isomerization and olefin exchange reactions mediated by [(MeN2N)Ni(alkyl)] complexes to show that the second scenario is true here.80
It was found that when octyl-Br was coupled to i-propyl-MgCl, n-undecane was formed in a 91% yield (Scheme 31). The n-propyl end-group in the product originated from the i-propyl group of the Grignard reagent. Thus, an isomerization occurred at the Ni center. The isomerization was then studied in a stoichiometric manner. Reaction of 25 with i-propyl-MgCl gave the isomerized Ni alkyl complex, [(MeNN2)Ni-Prn] (30) in a 75% yield (Scheme 31). The direct transmetalation product, [(MeNN2)Ni-Pri] (31), could not be observed. The most probable reaction pathway for the isomerization of 31 to 30 is first β-H elimination from 31 to form a Ni–H (32) and propene, which re-inserts to form the more stable isomer 30 (Scheme 31).
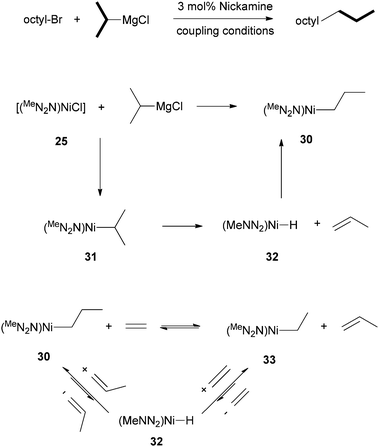 | ||
| Scheme 31 Study of β-H elimination from (N2N)Ni-alkyl complexes. | ||
To further verify the possibility of β-H elimination, olefin exchange experiments were carried out. Reaction of 30 with ethylene (5 to 20 bar) at 80 °C in benzene produced the Ni ethyl complex [(MeNN2)Ni–Et] (33) and 1-propene. The reaction is reversible and has an equilibrium constant of 0.68(3). Again, the reactions occur through first a β-H elimination to form a Ni–H complex, which inserts into one of the two olefins to form either the Ni-ethyl or Ni-methyl complex (Scheme 31).
The isomerization and olefin exchange experiment indicate that β-H elimination indeed occurs in [(MeN2N)Ni(alkyl)] complexes. However, several [(MeN2N)Ni(alkyl)] were isolated and they were stable in solution even when heated up to 80 °C. Thus, β-H elimination must be reversible and thermodynamically uphill. This was confirmed by DFT calculations, which showed that β-H elimination for 33 had an enthalpy change of 21.8 kcal mol−1. Indeed, the Ni hydride complex 32 was shown experimentally to react with ethylene to form 33.
This study reveals that the efficiency of Nickamine catalyst against unproductive β-H elimination has a thermodynamic origin. It suggests that to avoid β-H elimination, it is not necessary to enforce structural saturation at the metal center which may lead to lower activity for the desired reactions. If the ligands can induce unfavorable thermodynamics for β-H elimination, then a high efficiency can be obtained for cross coupling even with an open metal center. One can imagine the same scenario operates in reactions using other Ni catalysts presented in this Perspective.
6.3 Alkyl–aryl Kumada coupling using Nickamine as catalyst
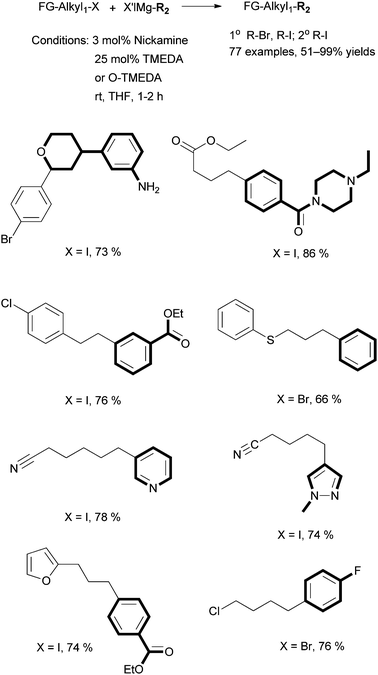 | ||
| Scheme 32 Alkyl–aryl Kumada coupling by Nickamine. | ||
When the coupling of octyl-I with PhMgCl was conducted in the presence of 100 equivalents of Hg (relative to the catalyst), octyl-Ph was produced in an 84% yield, nearly identical to the coupling in the absence of Hg. This suggests that heterogeneous metal particles are not responsible for the catalysis. In the coupling of octyl-I with PhMgBr, when 3 mol% of [(MeNN2)NiCl] (25) was used as the precatalyst, we isolated [(MeNN2)Ni–Ph] (34) in a 72% yield (relative to 1) from the catalysis mixture after acidic workup in air. Complex 34 was independently synthesized by reaction of 25 with PhMgBr, and it is stable under air and in protic solutions. A pre-made sample of 34 was used as the catalyst for the coupling of octyl-I and octyl-Br with PhMgBr. The reactions produced octyl-Ph in 92% and 72% yields, respectively. These results are comparable to those obtained using 25 as the catalyst. Thus, the Ni–Ph complex 34 is catalytically competent.
Interestingly, 34 failed to react with alkyl halides in stoichiometric reactions to give C–C coupled products. For example, the reaction of 34 with 20 equivalents of octyl-I under catalytically relevant conditions (in THF in the presence of TMEDA) was monitored by NMR, but no reaction was observed over 24 h (Scheme 33). Complex 34 did react with PhMgBr under the same conditions slowly to give a Mg complex [(MeNN2)(THF)Mg–Cl] (35), with a 25% conversion after 2 h (Scheme 33). The rate of this reaction is too slow to be relevant to the catalysis. 35 was shown not to be a competent catalyst for the coupling. However, when 34 was mixed with an equal mixture of PhMgCl, octyl-I, and TMEDA (all 1 equivalent relative to 34) in THF, a rapid reaction occurred, which gave the C–C coupled product octyl-Ph (Scheme 33).
![Reactivity of [(MeN2N)Ni-Ph].](/image/article/2011/SC/c1sc00368b/c1sc00368b-s33.gif) | ||
| Scheme 33 Reactivity of [(MeN2N)Ni-Ph]. | ||
Taking into consideration the reactivity and catalytic property of 34, we proposed that a Ni-(hetero)aryl species [(MeN2N)Ni-R2] (36, R2 is an aryl or heteroaryl group) is an intermediate for the alkyl–aryl coupling (Scheme 34). This intermediate is formed by transmetalation of the [(MeN2N)Ni–Cl] catalyst with a Grignard reagent. 36 reacts with a mixture of the alkyl halide and the Grignard reagent to give a six-coordinate complex [(MeN2N)Ni(alkyl)(R2)2] (37). Reductive elimination from 37 then gives the alkyl–aryl coupling product and regenerates 36. The high yield of the alkyl–aryl coupling suggests that alkyl–aryl reductive elimination is faster than aryl–aryl reductive elimination.
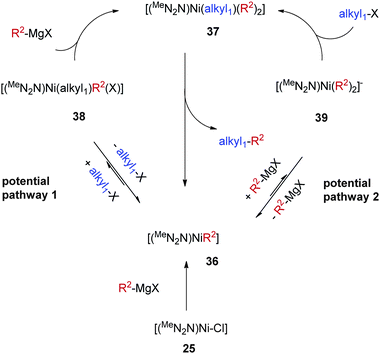 | ||
| Scheme 34 Catalytic cycle for alkyl–aryl Kumada coupling by Nickamine. | ||
From 36 to 37, there are two conceivable pathways. 36 could react with alkyl halide to form [(MeNN2)Ni(alkyl)(R2)X] (38), which could then be transmetalated by the Grignard reagent to give 37 (potential pathway 1, Scheme 34). Alternatively, 36 could react with a Grignard reagent to give an anionic species [(MeNN2)Ni(R2)2]− (39), which then reacts with the alkyl halide to form 37 (potential pathway 2, Scheme 34). Model reactions between 34 and octyl-I or PhMgCl did not produce 38 or 39 in detectable amounts (Scheme 33). This however does not exclude either pathway, as the transformation from 36 to 38 or 39 can be fast and reversible, but thermodynamically uphill. Thus 38 or 39 exists in a very small percentage, but upon treatment with a second coupling partner, quickly forms 37 and then the coupling product. This is supported by the fact that 34 reacted rapidly with a mixture of octyl-I and PhMgCl to give octyl-Ph (Scheme 33). It is tempting to favor pathway 1 in Scheme 34 due to the resemblance to the mechanism for alkyl–alkyl coupling (Scheme 30); however, pathway 2 cannot be easily ruled out, especially in light of the coupling chemistry developed by Kambe et al. (Scheme 6).
Like in complexes 28 and 29, the oxidation states of Ni in 37 and 38 are still uncertain. The Ni center can be Ni(IV), Ni(III), or Ni(II) depending on the oxidation states of the ligand. Ni(IV) is reasonable because the complex would then be an 18-e organometallic species. Yet one needs to prepare and study complexes of this type to learn more about their electronic structure.
6.4 Direct C–H functionalization using non-activated alkyl halides as electrophiles
Mechanistic studies indicate that organometallic nickel complexes [(MeN2N)Ni–R] are key intermediates for the cross coupling of alkyl halides. These intermediates are generated by transmetalation with organometallic nucleophiles, for example, Grignard reagents. For this reason, at least a stoichiometric amount of organometallic reagent is needed for cross coupling. The same principle applies to other types of coupling reactions such as Suzuki, Negishi, and Hiyama coupling. The use of organometallic reagents can be problematic due to concerns with cost, safety, and waste treatment.We hypothesized that if [(MeN2N)Ni–R] intermediates could be generated without a preformed organometallic reagent, a more benign coupling reaction would be developed. An interesting possibility is to generate the nucleophiles from C–H moieties of the substrates. We decided to first explore the direct alkylation of weakly acidic C–H bonds. These C–H bonds could be deprotonated by a weak base to give a small percentage or trace amount of carbon anions. If these carbon anions could be transferred to the [(MeN2N)Ni] fragment, the [(MeN2N)Ni–R] species would be generated. A Cu co-catalyst might be used to trap and transfer the carbon anion. Reactions of [(MeN2N)Ni–R] with an non-activated alkyl halide would lead to direct C–H alkylation. Following this design, we developed the coupling of non-activated alkyl halides with terminal alkynes and aromatic heterocycles using Nickamine as the pre-catalyst.
We reported in 2009 the first Ni-catalyzed coupling of non-activated alkyl halides with terminal alkynes (Scheme 35).87 The coupling of primary alkyl iodides was successful at 100 °C in dioxane using 5 mol% Nickamine (25) as (pre)catalyst, 3 mol% CuI as co-catalyst, and 1.4 equiv. of Cs2CO3 as base. To couple alkyl bromides, 20 mol% of NaI additive was required, presumably to convert alkyl bromides into iodides. To couple alkyl chlorides, 20 mol% of nBu4NI and a temperature of 140 °C were required. This was the first time that alkyl chlorides had been used in Sonogashira reactions. The catalysis tolerates a wide range of functional groups in both coupling partners. Substituted alkynes could also be prepared in comparable yields by reactions of alkyl halides with alkynyl anions in liquid ammonia solutions. However, the latter methods require a strong base, and thus have limited functional-group tolerance. Therefore, the Ni-catalyzed Sonogashira protocol is advantageous for the preparation of highly functionalized alkynes.
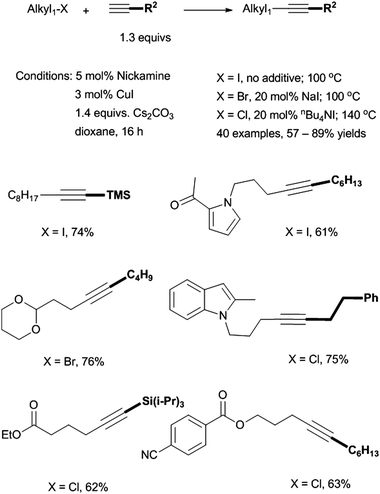 | ||
| Scheme 35 Sonogashira coupling of alkyl halides by Nickamine. | ||
One of the useful features in this Ni-catalyzed Sonogashira coupling is that it is selective among alkyl halides under certain reaction conditions. It is therefore possible to carry out sequential coupling reactions on substrates containing more than one type of alkyl–X bond (Scheme 36). Furthermore, the current protocol for Sonogashira coupling of alkyl chlorides can be combined with the Kumada coupling protocols for alkyl bromides and iodides, which leads to the multiple-functionalization of substrates with both alkyl–Cl and alkyl–Br or alkyl–I units (Scheme 36).
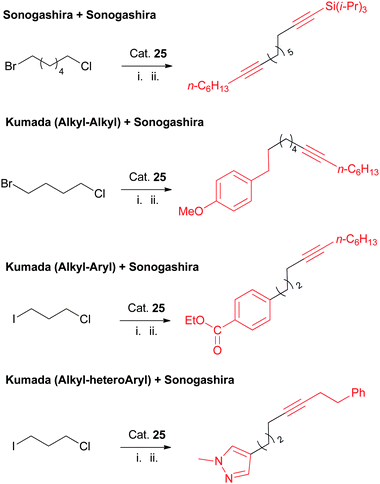 | ||
| Scheme 36 Sequential/orthogonal functionalization of alkyl halides using Nickamine catalyst. | ||
The mechanism of the catalysis has not been investigated in detail. Hg-test ruled out the possibility that the reactions were catalyzed by Ni(0) particles or colloids.79
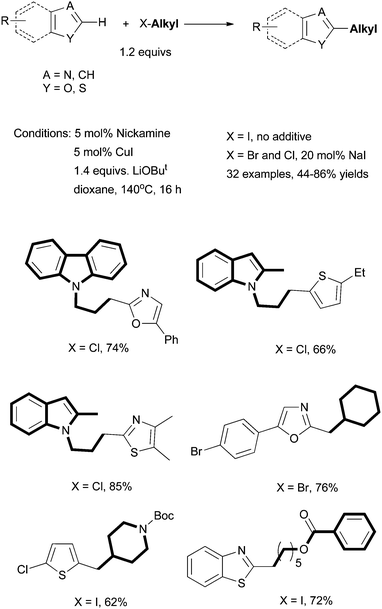 | ||
| Scheme 37 Direct alkylation of aromatic heterocycles using Nickamine as pre-catalyst. | ||
The catalytically active Ni-species was probed by NMR. During and after a coupling reaction, HMeN2N was detected as the only ligand-containing molecules. This result suggests that the Nickamine pre-catalyst degrades into another Ni-containing species under the catalytic conditions. The Hg-test showed that the yield of a coupling reaction decreased significantly in the presence of 100 equiv. of Hg (relative to Ni). If a reaction mixture was filtered to remove insoluble solids at a partial conversion, and the homogeneous filtrate solution was allowed to react further, the overall conversion and yield also diminished. These two results indicate that the reactions are catalyzed by nickel metal particles. It seems that Nickamine is the best pre-catalyst for generating the active Ni particles. Other soluble nickel complexes, e.g., [NiCl2(dme)] and [NiCl2(PPh3)] are not efficient pre-catalysts.
The mechanism of the catalysis remains speculative at the moment. The first step might be the deprotonation of the C(2)–H by a base to form a carbon anion. In the coupling of benzoxazole, the existence of anionic benzoxazolate was confirmed by a D2O quenching experiment. Almost no isotopic effect was observed, suggesting that the C–H cleavage is not the turnover-limiting step. The Cu co-catalyst facilitates the transmetalation of the carbon anion to nickel. The combination of transmetalation with oxidative addition of alkyl halides gives a Ni(II)(alkyl)(heteroAryl) species, which undergoes reductive elimination to give the coupling product and Ni particle.
7. Catalysis employing a bidentate amino-amide ligand
7.1 Ligand development
One limitation of the Nickamine-catalyzed Kumada coupling is the low efficiency on the coupling of secondary alkyl halides (see section 7.2). We suspected that this low efficiency was due to the steric encumbrance of the pincer ligand on a square-planar Ni(II) center. To largely maintain the electronic property of the pincer ligand while giving more structural flexibility to the metal center, we developed the bidentate mixed amino-amide ligands, Pengamine (Scheme 38).76,89 A series of Ni(II) complexes of these ligands were prepared. These complexes have different structures, coordination numbers, and spins. They were all characterized by X-ray crystallography.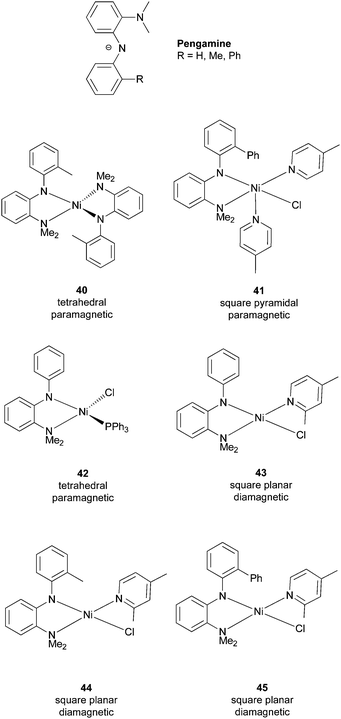 | ||
| Scheme 38 Pengamine ligands and their Ni complexes. | ||
7.2 Coupling of secondary alkyl halides
The Ni(II) complexes with Pengamine ligands were studied for their efficiency in the alkyl–alkyl Kumada coupling of non-activated secondary alkyl halides.89 The performance of the Nickamine catalyst (25) was used as a reference. The coupling of 2-iodobutane with n-octyl-MgCl and cyclohexyl iodide with n-butyl-MgCl were used as the test reactions (Scheme 39). The Nickamine catalyst was not efficient, giving a coupling yield of 4% and 46%, respectively. Complex 40 which has two bidentate ligands on Ni, was completely ineffective. It was shown earlier that transmetalation of the Ni–halide bond in Nickamine was a key step in alkyl–alkyl coupling. The lack of a Ni–halide bond might explain the inability of 40 to catalyze the coupling. Complexes 41–45 all have a Ni–halide bond, and indeed they all exhibit some degree of catalytic efficiency. Thus, a transmetalation site on the catalyst is essential in this type of coupling.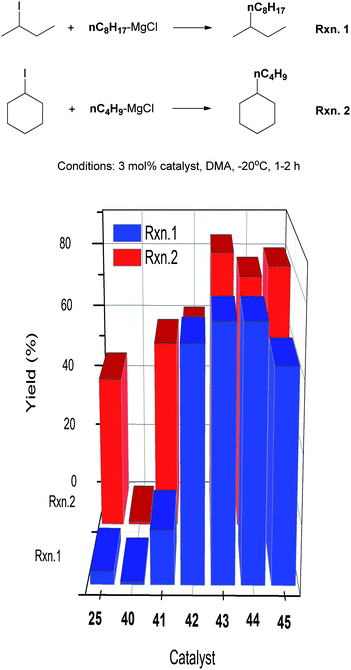 | ||
| Scheme 39 Structure–activity study of Ni-Pengamine catalysts for coupling of secondary alkyl halides. | ||
The four-coordinate complexes 42–45 are more efficient than the five-coordinate complex 41. Gratifyingly, catalysts 42–45 performed much better than Nickamine. Their higher efficiency was attributed to the fact that the 4th ligand in these complexes, namely, PPh3 or 2,4-lutidine, dissociates from the Ni center during catalysis to create a 3-coordinate complex which is more open. Ligand dissociation was confirmed by inhibition study which showed that addition of external ligands decreased the conversion and yield.89
Catalysts 42 and 43 were employed for the coupling of other secondary alkyl iodides and bromides (Scheme 40). Modest to good yields were achieved. In most cases, 43 was more efficient. Nevertheless, for some bulky acyclic alkyl iodides, 42 is a better catalyst. Among catalysts known for alkyl–alkyl Kumada coupling of non-activated secondary alkyl halides, these two complexes have the best scope.
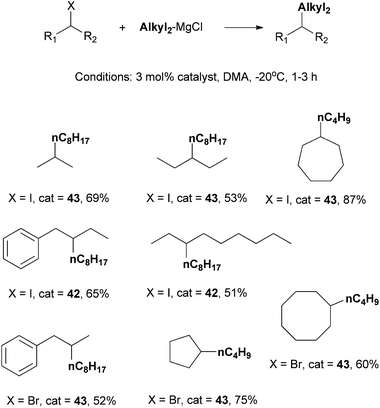 | ||
| Scheme 40 Scope of Kumada coupling of secondary alkyl halides using Ni-Pengamine catalysts. | ||
The mechanism of alkyl–alkyl coupling by Ni-Pengamine catalysts has not yet been studied in detail. A catalytic cycle analogous to the one in Scheme 30 is reasonable but needs to be probed experimentally. The reactions using radical-probe substrates indicate that the activation of secondary alkyl halides occurs via a radical process. The combination of secondary alkyl radical with the nickel center has a rate that is slower than the ring-closing rearrangement of the hept-6-en-2-radical, which has a first-order rate constant of about 105 s−1.
8. Summary and outlook
Significant progress has been made in Ni-catalyzed cross coupling of non-activated alkyl halides. Methods are now available for all main types of cross coupling reactions, namely Suzuki, Negishi, Stille, Hiyama, Sonogashira, and Kumada coupling. A growing number of ligands can be employed. Many catalytic systems exhibit a wide substrate scope and a high functional group tolerance. Thus, the Ni-catalysis is ready for preparative chemistry. Fu et al. applied Ni-catalyzed Negishi coupling for the formal synthesis of α-cembra-2,7,11-triene-4,6-diol63 and Fluvirucinine A1.49 Phillips et al. employed Pd-catalyzed alkyl–alkyl Suzuki coupling for the total synthesis of (+)-Spirolaxine methyl ether90 and Pyranicin.91 With the increasing awareness of the synthetic community of these new coupling methodologies, the cross coupling of non-activated alkyl halides is expected to become a standard tool in organic synthesis, benefiting the production of synthetic building blocks, fine and speciality chemicals, agro-chemicals, biologically and pharmaceutically active molecules, and organic materials.One of the most spectacular developments in this area is the asymmetric cross coupling of racemic alkyl halides. Initially only activated alkyl halides were used. In the last several years, however, Fu et al. have developed three methods for the coupling of non-activated alkyl halides.71–73 The substrates are still limited to those containing pendant aryl, nitrogen, or oxygen atoms. As the field is gaining momentum, one can anticipate exciting advances in the next years. Such advances may impact the way chiral molecules are made in the future.
Looking forward, the efficiency of Ni-catalysis remains to be improved. In all examples discussed herein, the loading of Ni catalyst is above 1 mol%, and often near 10 mol%. It is not easy to lower the catalyst loading, because the productive coupling catalysis has to compete with side reactions. Non-activated alkyl halides suffer from a variety of side reactions under coupling conditions, e.g., base-mediated HX elimination, halide–halide and halide–metal exchange reactions, and homo-coupling. A high loading is often necessary to increase the rate of cross coupling. For example, in the alkyl–alkyl Kumada coupling catalyzed by Nickamine, the catalyst itself does not decompose and can be isolated in a nearly quantitative yield after the reaction. However, 3 mol% catalyst is needed for cross coupling to out-compete side reactions.41 More active catalytic systems will be more attractive for industrial applications.
The mechanism of these Ni-catalyzed coupling reactions is complex and not fully understood. It appears that there are multiple reaction pathways, and the mechanism for a particular system depends on the ligands. Three alkyl–alkyl coupling systems have been studied, and the following proposals can be made, albeit with some uncertainty (Fig. 2). The common feature of the three systems is that transmetalation is required to form Ni alkyl species before oxidative addition can occur. When dienes are used as ligands, the catalysis occurs via a Ni(II)/Ni(IV) cycle, and the activation of alkyl halides is by nucleophilic substitution (Scheme 6). When imine and pyridine type ligands are used, the catalysis occurs via a Ni(II)−L−/Ni(II)/Ni(III)/Ni(I) cycle, and the ligands are involved in the redox chemistry (Scheme 20). When pincer bis(amino)amide is used as the ligand, the catalysis occurs via a LNiII(R1)/LNi(R1)X/LNi(R1)(R2)X/LNiIIX cycle (Scheme 30). The oxidation states of Ni centers in the intermediates are yet to be assigned. For the latter two classes of ligands, the activation of alkyl halides probably occurs via a radical-rebound mechanism. A radical chain mechanism is possible as well. Radical chain mechanism was suggested for the reaction of Ni-allyl complexes with organic halides back in 1975 because racemization and inhibition by radical scavengers were observed.92 However, these observations can also be explained by a radical-rebound mechanism. Kinetic studies can be used to distinguish these two mechanisms, and such studies are worthwhile future research topics.
 | ||
| Fig. 2 Ligand-dependent mechanistic features of Ni-catalyzed cross coupling of non-activated alkyl halides. | ||
The mechanistic understanding of Ni-catalyzed cross coupling of non-activated alkyl halides is essential for the further development of the field. One can use Pd-catalysis as an analogy. The success of Pd-catalyzed cross coupling as highly reliable and widely applicable chemistry can be attributed to the tremendous amount of mechanistic knowledge in Pd-mediated or catalyzed reactions. The mechanistic study is however challenging. In contrast to Pd, Ni has the tendency to mediate both one- and two-electron redox couples, which opens up more reaction pathways. Paramagnetic species are often involved, which complicates analysis. Ligand redox chemistry might be important, making it hard to assign the oxidation state of the metal. It is also difficult to identify the catalytically active species when mono- or bidentate ligands are used in conjunction with Ni salts in catalysis. As we found in the coordination chemistry of bidentate Pengamine ligands,89Ni complexes with different coordination number, geometry, spin state, and catalytic efficiency can be produced depending on the reaction condition. In this aspect, it is advantageous to use well-defined catalysts for mechanistic investigations.
Our work in Ni-catalysis is supported by the Swiss National Science Foundation (project no. 200021_126498) and the EPFL. I thank my coworkers for their experimental work and intellectual contributions. The credits are particularly due to Oleg Vechorkin, Zsolt Cosk, Peng Ren, and Jan Breitenfeld. I thank Prof. Jérôme Waser (EPFL) and Prof. Karl Gademann (Univ. Basel) for many helpful suggestions.
Notes and references
- A. Suzuki, Angew. Chem., Int. Ed., 2011, 50, 6722–6737 CrossRef CAS; E. Negishi, Angew. Chem., Int. Ed., 2011, 50, 6738–6764 CrossRef.
- A. de Meijere and F.Diederich, ed., Metal-Catalyzed Cross-Coupling Reactions, Wiley-VCH, Weinheim, 2004 Search PubMed.
- E. Negishi, ed., Handbook of Organopalladium Chemistry for Organic Synthesis, Wiley-InterScience, New York, 2002 Search PubMed.
- T. Y. Luh, M. K. Leung and K. T. Wong, Chem. Rev., 2000, 100, 3187–3204 CrossRef CAS.
- D. J. Cardenas, Angew. Chem., Int. Ed., 1999, 38, 3018–3020 CrossRef CAS.
- M. R. Netherton and G. C. Fu, Adv. Synth. Catal., 2004, 346, 1525–1532 CrossRef CAS.
- A. C. Frisch and M. Beller, Angew. Chem., Int. Ed., 2005, 44, 674–688 CrossRef CAS.
- A. Rudolph and M. Lautens, Angew. Chem., Int. Ed., 2009, 48, 2656–2670 CrossRef CAS.
- M. Tamura and J. Kochi, J. Am. Chem. Soc., 1971, 93, 1483–1485 CrossRef CAS; M. Tamura and J. Kochi, J. Am. Chem. Soc., 1971, 93, 1485–1487 CrossRef; M. Tamura and J. Kochi, J. Organomet. Chem., 1971, 31, 289–309 CrossRef.
- T. Ishiyama, S. Abe, N. Miyaura and A. Suzuki, Chem. Lett., 1992, 691–694 CrossRef CAS.
- A. Devasagayaraj, T. Studemann and P. Knochel, Angew. Chem., Int. Ed. Engl., 1995, 34, 2723–2725 CrossRef CAS.
- B. D. Sherry and A. Furstner, Acc. Chem. Res., 2008, 41, 1500–1511 CrossRef CAS; D. J. Cardenas, Angew. Chem., Int. Ed., 2003, 42, 384–387 CrossRef; F. Glorius, Angew. Chem., Int. Ed., 2008, 47, 8347–8349 CrossRef.
- J. Terao and N. Kambe, Acc. Chem. Res., 2008, 41, 1545–1554 CrossRef CAS.
- V. B. Phapale and D. J. Cádenas, Chem. Soc. Rev., 2009, 38, 1598–1607 RSC.
- F. A. Cotton, G. Wilkinson, C. A. Murillo and M. Bochmann, Advanced Inorganic Chemistry, Wiley-Interscience, 1999 Search PubMed.
- R. A. Gossage, L. A. Van De Kuil and G. Van Koten, Acc. Chem. Res., 1998, 31, 423–431 CrossRef CAS; V. Pandarus and D. Zargarian, Organometallics, 2007, 26, 4321–4334 CrossRef.
- T. J. Collins, T. R. Nichols and E. S. Uffelman, J. Am. Chem. Soc., 1991, 113, 4708–4709 CrossRef CAS.
- S. Mandal and E. S. Gould, Inorg. Chem., 1995, 34, 3993–3997 CrossRef CAS.
- G. D. Jones, J. L. Martin, C. McFarland, O. R. Allen, R. E. Hall, A. D. Haley, R. J. Brandon, T. Konovalova, P. J. Desrochers, P. Pulay and D. A. Vicic, J. Am. Chem. Soc., 2006, 128, 13175–13183 CrossRef CAS.
- D. Adhikari, S. Mossin, F. Basuli, J. C. Huffman, R. K. Szilagyi, K. Meyer and D. J. Mindiola, J. Am. Chem. Soc., 2008, 130, 3676–3682 CrossRef CAS.
- P. J. Chirik and K. Wieghardt, Science, 2010, 327, 794–795 CrossRef CAS.
- Fv. Lovecchi, E. S. Gore and D. H. Busch, J. Am. Chem. Soc., 1974, 96, 3109–3118 CrossRef.
- J. C. Fontecilla-Camps, P. Amara, C. Cavazza, Y. Nicolet and A. Volbeda, Nature, 2009, 460, 814–822 CrossRef CAS.
- S. Scheller, M. Goenrich, S. Mayr, R. K. Thauer and B. Jaun, Angew. Chem., Int. Ed., 2010, 49, 8112–8115 CrossRef CAS.
- C. M. Lee, C. H. Chen, F. X. Liao, C. H. Hu and G. H. Lee, J. Am. Chem. Soc., 2010, 132, 9256–9258 CrossRef CAS.
- V. Dimitrov and A. Linden, Angew. Chem., Int. Ed., 2003, 42, 2631–2633 CrossRef CAS.
- H. F. Klein, A. Bickelhaupt, T. Jung and G. Cordier, Organometallics, 1994, 13, 2557–2559 CrossRef CAS.
- W. Z. Chen, S. Shimada, M. Tanaka, Y. Kobayashi and K. Saigo, J. Am. Chem. Soc., 2004, 126, 8072–8073 CrossRef CAS.
- T. T. Tsou and J. K. Kochi, J. Am. Chem. Soc., 1979, 101, 6319–6332 CrossRef CAS.
- T. Yamamoto, A. Yamamoto and S. Ikeda, J. Am. Chem. Soc., 1971, 93, 3350–3359 CrossRef CAS.
- J. F. Hartwig, Organotransition Metal Chemistry-From Bonding to Catalysis, University Science Books, Sausalito, California, 2010 Search PubMed.
- R. Giovannini, T. Studemann, A. Devasagayaraj, G. Dussin and P. Knochel, J. Org. Chem., 1999, 64, 3544–3553 CrossRef CAS.
- R. Giovannini, T. Studemann, G. Dussin and P. Knochel, Angew. Chem., Int. Ed., 1998, 37, 2387–2390 CrossRef CAS.
- R. Giovannini and P. Knochel, J. Am. Chem. Soc., 1998, 120, 11186–11187 CrossRef CAS.
- M. Piber, A. E. Jensen, M. Rottlander and P. Knochel, Org. Lett., 1999, 1, 1323–1326 CrossRef CAS.
- A. E. Jensen and P. Knochel, J. Org. Chem., 2002, 67, 79–85 CrossRef CAS.
- J. Terao, H. Watanabe, A. Ikumi, H. Kuniyasu and N. Kambe, J. Am. Chem. Soc., 2002, 124, 4222–4223 CrossRef CAS.
- J. Terao, A. Ikumi, H. Kuniyasu and N. Kambe, J. Am. Chem. Soc., 2003, 125, 5646–5647 CrossRef CAS.
- J. Terao, Y. Naitoh, H. Kuniyasu and N. Kambe, Chem. Commun., 2007, 825–827 RSC.
- R. Martin and S. L. Buchwald, J. Am. Chem. Soc., 2007, 129, 3844–3845 CrossRef CAS.
- O. Vechorkin and X. L. Hu, Angew. Chem., Int. Ed., 2009, 48, 2937–2940 CrossRef CAS.
- S. P. Singh, J. Terao and N. Kambe, Tetrahedron Lett., 2009, 50, 5644–5646 CrossRef CAS.
- J. Terao, H. Todo, H. Watanabe, A. Ikumi and N. Kambe, Angew. Chem., Int. Ed., 2004, 43, 6180–6182 CrossRef CAS.
- J. Terao, H. Todo, S. A. Begum, H. Kuniyasu and N. Kambe, Angew. Chem., Int. Ed., 2007, 46, 2086–2089 CrossRef CAS.
- G. A. Chass, E. A. B. Kantchev and D. C. Fang, Chem. Commun., 2010, 46, 2727–2729 RSC.
- J. R. Zhou and G. C. Fu, J. Am. Chem. Soc., 2003, 125, 14726–14727 CrossRef CAS.
- C. Fischer and G. C. Fu, J. Am. Chem. Soc., 2005, 127, 4594–4595 CrossRef CAS.
- F. O. Arp and G. C. Fu, J. Am. Chem. Soc., 2005, 127, 10482–10483 CrossRef CAS.
- S. Son and G. C. Fu, J. Am. Chem. Soc., 2008, 130, 2756–2757 CrossRef CAS.
- P. M. Lundin, J. Esquivias and G. C. Fu, Angew. Chem., Int. Ed., 2009, 48, 154–156 CrossRef CAS; S. Lou and G. C. Fu, J. Am. Chem. Soc., 2010, 132, 1264–1266 CrossRef.
- S. W. Smith and G. C. Fu, J. Am. Chem. Soc., 2008, 130, 12645–12647 CrossRef CAS.
- H. G. Gong and M. R. Gagné, J. Am. Chem. Soc., 2008, 130, 12177–12183 CrossRef CAS.
- J. Caeiro, J. P. Sestelo and L. A. Sarandeses, Chem.–Eur. J., 2008, 14, 741–746 CrossRef CAS.
- S. M. Goldup, D. A. Leigh, R. T. McBurney, P. R. McGonigal and A. Plant, Chem. Sci., 2010, 1, 383–386 RSC.
- V. B. Phapale, E. Bunuel, M. Garcia-Iglesias and D. J. Cádenas, Angew. Chem., Int. Ed., 2007, 46, 8790–8795 CrossRef CAS.
- X. L. Yu, T. Yang, S. L. Wang, H. L. Xu and H. G. Gong, Org. Lett., 2011, 13, 2138–2141 CrossRef CAS.
- J. Zhou and G. C. Fu, J. Am. Chem. Soc., 2004, 126, 1340–1341 CrossRef CAS.
- D. A. Powell and G. C. Fu, J. Am. Chem. Soc., 2004, 126, 7788–7789 CrossRef CAS.
- D. A. Powell, T. Maki and G. C. Fu, J. Am. Chem. Soc., 2005, 127, 510–511 CrossRef CAS.
- D. A. Everson, R. Shrestha and D. J. Weix, J. Am. Chem. Soc., 2010, 132, 920–921 CrossRef CAS.
- T. J. Anderson, G. D. Jones and D. A. Vicic, J. Am. Chem. Soc., 2004, 126, 8100–8101 CrossRef CAS.
- G. D. Jones, C. McFarland, T. J. Anderson and D. A. Vicic, Chem. Commun., 2005, 4211–4213 RSC.
- S. W. Smith and G. C. Fu, Angew. Chem., Int. Ed., 2008, 47, 9334–9336 CrossRef CAS.
- M. R. Prinsell, D. A. Everson and D. J. Weix, Chem. Commun., 2010, 46, 5743–5745 RSC.
- X. F. Lin and D. L. Phillips, J. Org. Chem., 2008, 73, 3680–3688 CrossRef CAS.
- F. Gonzalez-Bobes and G. C. Fu, J. Am. Chem. Soc., 2006, 128, 5360–5361 CrossRef CAS.
- N. A. Strotman, S. Sommer and G. C. Fu, Angew. Chem., Int. Ed., 2007, 46, 3556–3558 CrossRef CAS.
- B. Saito and G. C. Fu, J. Am. Chem. Soc., 2007, 129, 9602–9603 CrossRef CAS.
- Z. Lu and G. C. Fu, Angew. Chem., Int. Ed., 2010, 49, 6676–6678 CrossRef CAS.
- X. Dai, N. A. Strotman and G. C. Fu, J. Am. Chem. Soc., 2008, 130, 3302–3303 CrossRef CAS.
- B. Saito and G. C. Fu, J. Am. Chem. Soc., 2008, 130, 6694–6695 CrossRef CAS.
- N. A. Owston and G. C. Fu, J. Am. Chem. Soc., 2010, 132, 11908–11909 CrossRef CAS.
- L. Z., A. Wilsily and G. C. Fu, J. Am. Chem. Soc., 2011, 133, 8154–8157 CrossRef.
- Z. Csok, O. Vechorkin, S. B. Harkins, R. Scopelliti and X. L. Hu, J. Am. Chem. Soc., 2008, 130, 8156–8157 CrossRef CAS.
- O. Vechorkin, Z. Csok, R. Scopelliti and X. L. Hu, Chem.–Eur. J., 2009, 15, 3889–3899 CrossRef CAS.
- In our lab, Zsolt Csok and Oleg Vechorkin developed the synthesis of Lockamine ligand, and Peng Ren developed the synthesis of Pengamine ligands.
- P. Ren, O. Vechorkin, Z. Csok, I. Salihu, R. Scopelliti and X. L. Hu, Dalton Trans., 2011 10.1039/c1031dt10195a.
- Complex 25 is now commercially available from Sigma-Aldrich bearing the synonym ‘Nickamine’. The name reflects the combination of nickel and amine ligands.
- O. Vechorkin, Ph.D. Thesis EPFL, Lausanne, 2011.
- J. Breitenfeld, O. Vechorkin, C. Corminboeuf, R. Scopelliti and X. L. Hu, Organometallics, 2010, 29, 3686–3689 CrossRef CAS.
- O. Vechorkin, V. Proust and X. L. Hu, J. Am. Chem. Soc., 2009, 131, 9756–9766 CrossRef CAS.
- P. Knochel, A. Krasovskiy and I. Sapountzis, in Handbook of Functionalized Organometallics, ed. P. Knochel, Wiley-VCH, Weinheim, 2005, pp. 109–172 Search PubMed.
- R. I. Yousef, B. Walfort, T. Ruffer, C. Wagner, H. Schmidt, R. Herzog and D. Steinborn, J. Organomet. Chem., 2005, 690, 1178–1191 CrossRef CAS.
- A. Krasovskiy, C. Duplais and B. H. Lipshutz, J. Am. Chem. Soc., 2009, 131, 15592–15593 CrossRef CAS.
- M. Eckhardt and G. C. Fu, J. Am. Chem. Soc., 2003, 125, 13642–13643 CrossRef CAS.
- G. Altenhoff, S. Wurtz and F. Glorius, Tetrahedron Lett., 2006, 47, 2925–2928 CrossRef CAS.
- O. Vechorkin, D. Barmaz, V. Proust and X. L. Hu, J. Am. Chem. Soc., 2009, 131, 12078–12079 CrossRef CAS.
- O. Vechorkin, V. Proust and X. L. Hu, Angew. Chem., Int. Ed., 2010, 49, 3061–3064 CrossRef CAS.
- P. Ren, O. Vechorkin, K. von Allmen, R. Scopelliti and X. L. Hu, J. Am. Chem. Soc., 2011, 133, 7084–7095 CrossRef CAS.
- K. A. Keaton and A. J. Phillips, Org. Lett., 2007, 9, 2717–2719 CrossRef CAS.
- N. D. Griggs and A. J. Phillips, Org. Lett., 2008, 10, 4955–4957 CrossRef CAS.
- L. S. Hegedus and L. L. Miller, J. Am. Chem. Soc., 1975, 97, 459–460 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2011 |