Synthesis of human GLP-1 (7–36) by chemoselective α-ketoacid–hydroxylamine peptide ligation of unprotected fragments†
Jian
Wu
a,
Javier
Ruiz-Rodríguez
c,
Jeanne M.
Comstock
b,
Jesse Z.
Dong
b and
Jeffrey W.
Bode
*c
aRoy and Diana Vagelos Laboratories, Department of Chemistry, University of Pennsylvania, Philadelphia, PA 19104-6323, USA
bIPSEN/Biomeasure, Inc., 27 Maple Street, Milford, MA, USA
cLaboratorium für Organische Chemie, ETH–Zürich, Zürich, 8093, Switzerland. E-mail: bode@org.chem.ethz.ch; Tel: +41 44 633 21 03
First published on 1st August 2011
Abstract
The synthesis of the bioactive form of human glucagon-like peptide, GLP-1 (7–36), by the chemoselective ligation of two unprotected fragments, a C-terminal peptide α-ketoacid with an N-terminal peptide hydroxylamine, is reported. No reagents are required for the ligation and no byproducts are produced. Unprotected glutamic acid, histidine, arginine, lysine, serine, and tyrosine residues do not interfere with the ligation; no side products arising from undesired reactions of the side chains are detected. This synthesis is the first reported example of the application of the α-ketoacid–hydroxylamine amide ligation to the synthesis of a complex, unprotected peptide. Its success underscores the potential broad utility of this amide ligation for the synthesis of complex peptides and related targets.
Introduction
Synthetic methods for the chemoselective union of two fully unprotected peptide fragments are highly prized for their ability to provide access to complex, functionally rich peptides and proteins without the need for extensive purification or subsequent chemical manipulations.1 In particular, the native chemical ligation of C-terminal peptide thioesters and peptides containing N-terminal cysteines has transformed the chemical synthesis and semi-synthesis of proteins.2In our own efforts we have recently recognized that C-terminal peptide α-ketoacids and N-terminal peptide hydroxylamines (KAHA ligation) undergo a chemoselective, decarboxylative condensation to give native peptide bonds.3 This reaction proceeds in polar solvents, does not require reagents, tolerates all unprotected functional groups commonly found in peptide side-chains, and produces only carbon dioxide as a byproduct. The potential to use this ligation reaction at a variety of C- and N-terminal ligation sites offers a means of significantly expanding the repertoire of known methods to selectively join unprotected molecules under mild, reagent free conditions. The ligation of short unprotected peptide fragments has been established by our group3,4 and others,5 but its extension to larger peptides of contemporary interest has not been tested. In this article, we establish that the KAHA ligation is suitable for the coupling of two fully unprotected peptide fragments by the synthesis of human GLP-1(7–36)-NH2(1), a 30-mer peptide (Scheme 1).
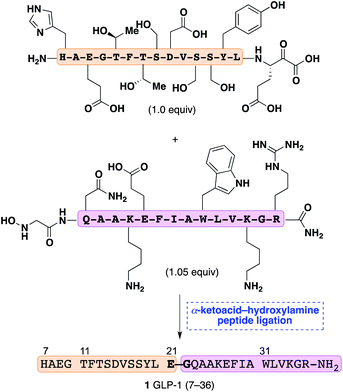 | ||
| Scheme 1 Synthesis of GLP-1 (7–36) by chemoselective α-ketoacid-hydroxylamine ligation of unprotected fragments. | ||
Despite the unparalleled ability of the native chemical ligation to form backbone amide bonds between two completely unprotected peptide or protein fragments under dilute aqueous conditions, limitations including the need for sulphur containing functionalities and challenging preparation of C-terminal thioesters6 have prompted the search for other general, yet equally selective, amide-forming ligations. The Staudinger ligation, reported independently by Bertozzi7 and Raines,8 is a promising approach but has not yet been demonstrated to ligate completely unprotected peptide fragments; however, exciting applications in protein immobilization and macrocyclization have appeared.9 A number of promising amide-forming reactions from thioacids and either azides or N-sulfonylamides have recently been studied as potential peptide ligations.10 These reactions currently require the protection of nucleophilic side-chains including lysine and cysteine, but have promise for certain synthetic applications such as the preparation of glycopeptides. Finally, the great importance and need for new peptide ligations has encouraged several groups to search for new amide-forming reactions. Towards this end, Danishefsky has reported an intriguing approach to amide formation by the reaction of isonitriles and thioacids11 and Johnston a remarkable oxidative amide-forming coupling of alpha-bromo nitriles and amines.12 Neither of these processes, unfortunately, are likely to tolerate unprotected amines.
Chemically synthesized proteins can often accommodate the incorporation of a cysteine or sulphur-containing auxiliary but such mutations are generally prohibited for therapeutic peptides. The KAHA-ligation could therefore serve as a valuable tool for the synthesis and eventual production of therapeutic peptides. To test its potential for this purpose we targeted the preparation of glucagon-like peptide 1 (GLP-1), a peptide hormone synthesized in intestinal endocrine cells.13 The biologically active form is widely recognized to be the 30-residue peptide comprised of residues 7–36 of proglucogen.14 Therapeutic applications include diabetes and obesity and two analogues, extenetide and liraglutide, are already approved for sale and a third, taspoglutide, has recently shown very promising Phase III results. As with the large number of other therapeutic peptides currently in use or in clinical trials, the challenge for their manufacture lies in producing the peptide in high purity at a reasonable cost under reproducible conditions.15,16 Stepwise syntheses of longer peptides often necessitate difficult purifications of the final product to remove deletion sequences or materials containing residual protecting groups.
Results and discussion
Preparation of the C-terminal α-ketoacid fragment
The preparation of C-terminal α-ketoacid fragment 2 began with the synthesis of cyanosulphurylide dipeptide 7 from protected glutamic acid (Scheme 2).4a With eventual large scale synthesis in mind, we chose to use this strategy rather than the sulphurylide linker we have previously reported.4b In order to avoid complications arising from Fmoc-deprotection of the side chain linked glutamic acid on 2-Cl-trityl resin, we prepared dipeptide 7 in solution phase by a straightforward approach. The loading of 7 onto commercially available 2-chlorotrityl resin proceeded under typical conditions. Subsequent elongation to the 15-residue fragment was carried out using standard Fmoc-based peptide synthesis on a commercial peptide synthesizer (Scheme 3). Resin cleavage and side chain deprotection with TFA gave the crude peptide without difficulties. Preparative HPLC provided pure 10 in good yield.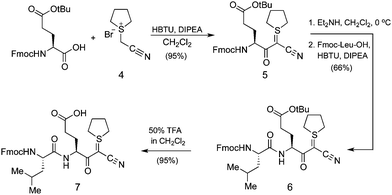 | ||
| Scheme 2 Synthesis of dipeptide cyanosulfurylide 7. | ||
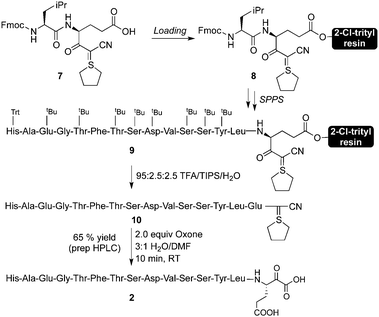 | ||
| Scheme 3 Synthesis of GLP-1 (7–21) α-ketoacid. | ||
In order to confirm that the cyanosulphurylide is configurationally stable during the solid phase peptide synthesis and resin cleavage, we performed the following experiment: unprotected peptide 10 was digested with trypsin to give three fragments, one of which was identified as 11 (Fig. 1). Three of the four possible stereoisomers of this fragment were prepared by solution phase chemical synthesis and isolated by analytical HPLC. Comparison of these standards to 11 obtained by trypsin digestion of 10 confirmed that no epimerization of the sulphur ylide occurred during any stage of its preparation or handling.
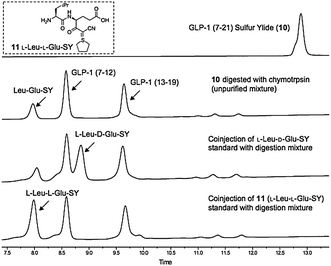 | ||
| Fig. 1 HPLC analysis of enzymatic cleavage of GLP-1 (7–21) sulfur ylide 10 (samples taken directly from the reaction mixture without purification or workup). | ||
Conversion of cyanosulphurylide 10 to the requisite α-ketoacid required a chemoselective oxidation in the presence of unprotected histidine, tyrosine, serine, threonine, and glutamic acid residues (Scheme 3). We were pleased to find that a slight modification, for solubility reasons, of our previously reported sulphur ylide oxidation4a using aqueous, acidic Oxone delivered the desired α-ketoacid with the formation of the corresponding C-terminal acid as the sole side product. Other oxidants, such as freshly prepared DMDO also delivered the α-ketoacid cleanly.
Unpurified ketoacid 2 could be used directly for ligation with hydroxylamine 3, but we found it advantageous to purify α-ketoacid 2 by preparative HPLC followed by lyophilization prior to ligation.
Synthesis of hydroxylamine fragment 3
Synthesis of the N-terminal hydroxylamine fragment commenced from fully protected, Rink Amide MBHA resin-linked peptide 12, which was prepared in modest purity on a commercial peptide synthesizer (Scheme 4). Although the peptide synthesis using this method could be further optimized, for the purposes of this study we utilized this material as we found (vide supra) that it was suitable for preparing pure N-terminal hydroxylamine 3.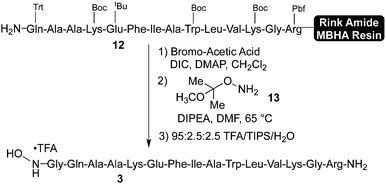 | ||
| Scheme 4 Synthesis of GLP-1 (22–36) hydroxylamine. | ||
In order to prepare the N-terminal glycine hydroxylamine, resin bound peptide 12 was first coupled with α-bromo acetic acid under standard conditions. This resulting material was treated with 10 equiv. of protected hydroxylamine 1317 in DMF at 65 °C. During these two steps, care must be taken to exclude water, which leads to the formation of an N-terminal α-hydroxyacetyl peptide. Resin cleavage and side chain deprotection under standard conditions afforded crude hydroxylamine 3, which was purified by preparative HPLC to afford pure 3 in moderate overall yield. The precise yield of this transformation is difficult to calculate due to the modest purity of the resin bound fragment 12, but we reproducibly obtained sufficient material following resin cleavage and purification. Alternatively, we have also found that hydroxylamine 3 can be prepared in solution by direct displacement of the crude N-terminal bromoacetyl peptide with hydroxylamine hydrochloride (see Supporting Information†). Hydroxylamine 3, which contains four basic residues in addition to the hydroxylamines, could not be purified on most standard HPLC columns and required columns with a densely bonded matrix.
Synthesis of GLP-1 by KAHA ligation
For solubility reasons, we selected a mixture of DMA and DMSO for the chemoselective ligation of α-ketoacid 2 and hydroxylamine 3. In our experience with other peptide ligations, reaction temperatures of approximately 40 °C were ideal. In this application, however, we found it necessary to warm the reaction to 60 °C for conversion, partly due to solubility considerations.18 The ligation was clean and rapid; proceeding nearly to completion after 4 h using only 1.05 equiv. of the hydroxylamine at only 10 mM (Fig. 2). The ligated peptide 1 was routinely obtained in 51% isolated yield following preparative HPLC, along with 12% of the D-Glu21 epimer.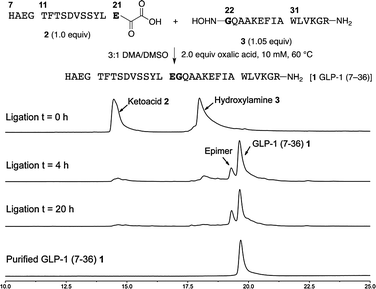 | ||
| Fig. 2 Synthesis of GLP-1 (7–36) 1 by chemoselective ligations of GLP-1 (7–21) α-ketoacid 2 and GLP-1 (22–36) hydroxylamine 3 and monitoring of the ligation by HPLC (samples taken directly from the reaction mixture without purification or workup). | ||
Extensive efforts to identify the origin of the epimerization pinpoint the oxidation of sulphur ylide 10 to α-ketoacid 2 as the culprit. The stereochemical integrity of sulfur ylide 10 had already been established by enzymatic degradation (see Fig. 1). Epimerization of the α-ketoacid during HPLC purification or the ligation was excluded by an experiment in which a ligation reaction was allowed to proceed to 30% completion before the unreacted α-ketoacid was recovered, isolated by preparative HPLC and resubjected to the ligation. Identical results were obtained; no additional epimerization was detected demonstrating that the α-ketoacid is stable to the ligation conditions and to purification by preparative HPLC. This is consistent with our experience with other α-ketoacids under acidic conditions. While we typically observe only trace amounts of epimerization during the oxidation of the sulphur ylide, we have established that it can occur during this process due to the configurational instability of the intermediate acyl cyanide. In this particular case, possibly due to the neighbouring unprotected carboxylic acid in the C-terminal glutamic acid residue, this intermediate epimerizes to a greater extent. An alternative approach to α-ketoacids is under development and should address this issue in the future.19
Conclusions
From these studies and other ongoing efforts in our laboratory, we can draw the following conclusions about the suitability of the α-ketoacid–hydroxylamine amide formation for chemoselective ligation of medium sized α-peptide fragments. 1) The reaction is exceptionally chemoselective. In no case have we detected the formation of any side-products arising from competing reactions of peptide side chains with either the hydroxylamine or the α-ketoacid. The current studies confirm that unprotected histidine, serine, glutamic acids, lysine, arginine, tyrosine, and tryptophan do not interfere with the ligation. Cysteine was not included in this study, but we have found that it is tolerated in parallel work. 2) The ligation reactions work with an equimolar stoichiometry of the α-ketoacids and hydroxylamine fragments and under dilute reaction conditions (10–20 mM).20 3) With hydroxylamines of the general structure R–NH–OH, the ligations work best in aprotic polar solvents such as DMF, DMA, or DMSO. Water is tolerated but with diminished reaction rates. In contrast, water is the preferred solvent for hydroxylamines of the structure R–NH–OBz or certain cyclic hydroxylamines.3b In these cases, however, higher reactant concentrations are usually required. 4) Reaction temperatures of 40–60 °C are generally preferred.Although further work is needed to improve the ligation conditions and suppress epimerization during the formation of the α-ketoacid, these studies establish the α-ketoacid–hydroxylamine peptide ligation as the first fully chemoselective ligation method for α-peptides that does not employ a C-terminal thioester. As methods for accessing and suitably protecting the requisite α-ketoacids and hydroxylamine functional groups improve,21 we believe that this reaction will develop into a robust, powerful approach to the chemoselective synthesis of therapeutic peptides, glycopeptides,22 peptidomimetics, and eventually proteins.
Acknowledgements
We thank IPSEN/Biomeasure and the NIH-NIGMS R01GM-076320 for support of this work. J. W. B. is a fellow of the Beckman Foundation, the Sloan Foundation, and the Packard Foundation.Notes and references
- For recent reviews of peptide ligations, see: (a) P. E. Dawson and S. B. H. Kent, Annu. Rev. Biochem., 2000, 69, 923–960 CrossRef CAS; (b) D. Macmillan, Angew. Chem., Int. Ed., 2006, 45, 7668–7672 CrossRef CAS; (c) C. P. R. Hackenberger and D. Schwarzer, Angew. Chem., Int. Ed., 2008, 47, 10030–10074 CrossRef CAS; (d) S. B. H. Kent, Chem. Soc. Rev., 2009, 38, 338–351 RSC.
- (a) P. E. Dawson, T. W. Muir, I. Clark-Lewis and S. B. H. Kent, Science, 1994, 266, 776 CAS; (b) B. L. Pentelute, Z. P. Gates, J. L. Dashnau and S. B. H. Kent, J. Am. Chem. Soc., 2008, 130, 9702–9707 CrossRef CAS; (c) Y. Sohma, B. L. Pentelute, J. Whittaker, Q. Hua and S. B. H. Kent, Angew. Chem., Int. Ed., 2008, 47, 1102–1106 CrossRef CAS.
- (a) J. W. Bode, R. M. Fox and K. D. Baucom, Angew. Chem., Int. Ed., 2006, 45, 1248–1252 CrossRef CAS; (b) N. Carrillo, E. A. Davalos, J. A. Russak and J. W. Bode, J. Am. Chem. Soc., 2006, 128, 1452–1453 CrossRef CAS.
- (a) L. Ju, A. R. Lippert and J. W. Bode, J. Am. Chem. Soc., 2008, 130, 4253–4255 CrossRef CAS; (b) L. Ju and J. W. Bode, Org. Biomol. Chem., 2009, 7, 2259–2264 RSC.
- (a) C. Cho, J. Liu, C. Chien, J. Shie, Y. Chen and J. Fang, J. Org. Chem., 2009, 74, 1549–1556 CrossRef CAS; (b) A. K. Sanki, R. S. Talan and S. J. Sucheck, J. Org. Chem., 2009, 74, 1886–1896 CrossRef CAS.
- For recent advances in peptide thioester synthesis, see: (a) A. Sewing and D. Hilvert, Angew. Chem., Int. Ed., 2001, 40, 3395–3396 CrossRef; (b) Y. Shin, K. A. Winans, B. J. Backes, S. B. H. Kent, J. A. Ellman and C. R. Bertozzi, J. Am. Chem. Soc., 1999, 121, 11684–11689 CrossRef CAS; (c) J. Brask, F. Albericio and K. J. Jensen, Org. Lett., 2003, 5, 2951–2953 CrossRef CAS; (d) J. B. Blanco-Canosa and P. E. Dawson, Angew. Chem., Int. Ed., 2008, 47, 6851–6855 CrossRef CAS; (e) G.-M. Fang, Y.-M. Li, F. Shen, Y.-C. Huang, J.-B. Li, Y. Lin, H.-K. Cui and L. Liu, Angew. Chem., Int. Ed., 2011, 50, 1–6 CrossRef; (f) R. Raz and J. Rademann, Org. Lett., 2011, 13, 1066–1609 CrossRef; (g) F. Mende and O. Seitz, Angew. Chem., Int. Ed., 2011, 50, 1232–1240 CrossRef CAS.
- E. Saxon, J. I. Armstrong and C. R. Bertozzi, Org. Lett., 2000, 2, 2141–2143 CrossRef CAS.
- (a) B. L. Nilsson, L. L. Kiessling and R. T. Raines, Org. Lett., 2000, 2, 1939–1941 CrossRef CAS; (b) B. L. Nilsson, L. L. Kiessling and R. T. Raines, Org. Lett., 2001, 3, 9–12 CrossRef CAS; (c) A. Tam, M. B. Soellner and R. T. Raines, J. Am. Chem. Soc., 2007, 129, 11421–11430 CrossRef CAS.
- (a) M. B. Soellner, K. A. Dickson, B. L. Nilsson and R. T. Raines, J. Am. Chem. Soc., 2003, 125, 11790–11791 CrossRef CAS; (b) J. Kalia, N. L. Abbott and R. T. Raines, Bioconjugate Chem., 2007, 18, 1064–1069 CrossRef CAS; (c) R. Kleineweischede and C. P. R. Hackenberger, Angew. Chem., Int. Ed, 2008, 47, 5984–5988 CAS.
- (a) N. Shangguan, S. Katukojvala, R. Greenberg and L. J. Williams, J. Am. Chem. Soc., 2003, 125, 7754–7755 CrossRef CAS; (b) D. Crich, K. Sana and S. Guo, Org. Lett., 2007, 9, 4423–4426 CrossRef CAS; (c) D. Crich, K. Sasaki, Md. Y. Rahaman and A. A. Bowers, J. Org. Chem., 2009, 74, 3886–3893 CrossRef CAS; (d) D. Crich and I. Sharma, Angew. Chem., Int. Ed., 2009, 48, 7591–7594 CrossRef CAS.
- (a) X. Li and S. J. Danishefsky, J. Am. Chem. Soc., 2008, 130, 5446 CrossRef CAS; (b) Y. Yuan, J. Zhu, X. Li, X. Wu and S. J. Danishefsky, Tetrahedron Lett., 2009, 50, 2329–2333 CrossRef CAS; (c) J. L. Storkdill, X. Wu and S. J. Danishefsky, Tetrahedron Lett., 2009, 50, 5152–5155 CrossRef.
- B. Shen, D. M. Makley and J. N. Johnston, Nature, 2010, 465, 1027–1032 CrossRef CAS.
- K. Dungan and J. B. Buse, Clinical Diabetes, 2005, 23, 56–62 CrossRef.
- (a) D. K. Arulmozhi and B. Portha, Eur. J. Pharm. Sci, 2008, 96–108 Search PubMed; (b) M. Rizzo, A. A. Rizvi and G. A. Spinas, Expert Opin. Invest. Drugs, 2009, 18, 1495–1503 CrossRef CAS.
- For synthetic studies of a GLP-1 analogue, see: J. Krüger, T. Minuth, W. Schröder and J. Werwath, Eur. J. Org. Chem., 2008, 5936–5945 CrossRef.
- (a) P. W. Latham, Nat. Biotechnol., 1999, 17, 755–757 CrossRef CAS; (b) A. M. Thayer, C&EN, 2011, 89, 21–25 Search PubMed.
- K. Mori and K. Koseki, Tetrahedron, 1988, 44, 6013–6020 CrossRef CAS.
- These peptide fragments also have appreciable solubility in the buffered system typically used in native chemical ligation (6 M guanidinium·HCl, 0.1 M phosphate buffer). Unfortunately the KAHA ligation did not proceed under these conditions.
- M. A. Flores and J. W. Bode, Org. Lett., 2010, 12, 1924–1927 CrossRef CAS.
- Higher reactant concentrations are not detrimental to the ligation but are rarely possible with medium-sized peptide fragments. This concentration range is comparable to the preferred conditions for native chemical ligation.
- For new procedures for the synthesis of peptide hydroxylamines, see: (a) T. Fukuzumi and J. W. Bode, J. Am. Chem. Soc., 2009, 131, 3864–3865 CrossRef CAS; (b) S. I. Medina, J. Wu and J. W. Bode, Org. Biomol. Chem., 2010, 8, 3405–3417 RSC.
- For recent advances in glycopeptide synthesis using chemical ligation, see: (a) Z. P. Tan, S. Y. Shang, T. Halkina, Y. Yuan and S. J. Danishefsky, J. Am. Chem. Soc., 2009, 131, 5424–5431 CrossRef CAS; (b) Y. Yuan, J. Chen, Q. Wan, Z. P. Tan, G. Chen, C. Kan and S. J. Danishefsky, J. Am. Chem. Soc., 2009, 131, 5432–5437 CrossRef CAS; (c) R. J. Payne and C. H. Wong, Chem. Commun., 2010, 46, 21–43 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c1sc00398d |
| This journal is © The Royal Society of Chemistry 2011 |