A flexible metal azolate framework with drastic luminescence response toward solvent vapors and carbon dioxide†
Xiao-Lin
Qi
,
Rui-Biao
Lin
,
Qing
Chen
,
Jian-Bin
Lin
,
Jie-Peng
Zhang
* and
Xiao-Ming
Chen
MOE Key Laboratory of Bioinorganic and Synthetic Chemistry, State Key Laboratory of Optoelectronic Materials and Technologies, School of Chemistry and Chemical Engineering, Sun Yat-Sen University, Guangzhou, 510275, China. E-mail: zhangjp7@mail.sysu.edu.cn
First published on 17th August 2011
Abstract
1H-Imidazo[4,5-f][1,10]phenanthroline (Hip) can be deprotonated to bridge Zn(II) ions, giving a flexible porous metal azolate framework [Zn7(ip)12](OH)2 (MAF-34) which shows not only drastic structural and luminescent changes for different solvent vapors, but also strong adsorption and quantitative luminescence response for low-pressure CO2.
Introduction
Porous coordination polymers (PCPs) are of great current interest because of their intriguing topologies and properties such as guest-responsive behaviors.1 For example, flexible or luminescent PCPs are potential sensors for small molecules.2 Metal azolate frameworks (MAFs) are an interesting subset of coordination polymers in the aspects of crystal engineering and functional materials.3 Especially, a series of zeolitic structures have been constructed by imidazolate derivatives.4 Nevertheless, studies in metal imidazolate frameworks are mainly focused on storage and separation properties based on their robust structures, whereas flexible, luminescent, or guest-responsive porous metal imidazolate frameworks remain scarcely reported.5 These imidazolate-based adsorbents also show potential for CO2 capture,4d but their performances are usually limited by the lack of strong CO2 binding sites.Generally, large π-conjugation systems and polynuclear metal clusters are responsible for the photoluminescence of coordination complexes.6 On the other hand, bulky molecular building blocks are disadvantageous for constructing porous structures. Therefore, luminescent PCPs are usually constructed by long bridging ligands with large aromatic rings. Nevertheless, the bridging length of imidazolate is short and unchangeable. Because metal imidazolate frameworks are based on single metal ions, and the π-conjugation system of commonly used imidazolate ligands are also small, they can only exhibit very low visible photoluminescence.
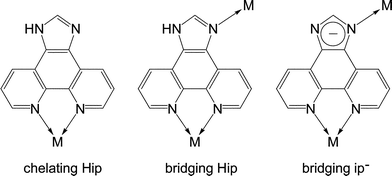
1H-Imidazo[4,5-f][1,10]phenanthroline (Hip) has been well studied for constructing luminescent complexes for interacting with DNA, but it is only used as a chelating ligand as a derivative of 2,2′-bipyridine and 1,10-phenathroline, keeping its imidazole moiety uncoordinated.7 Herein, we report a flexible PCP using the deprotonated 1H-imidazo[4,5-f][1,10]phenanthroline (ip−) as a bridging ligand, which shows not only a rare topological structure but also interesting structural and luminescent response toward different guest molecules including CO2.
Experimental section
General information
The ligand Hip was synthesized according to the literature method.8 Other reagents and solvents were commercially purchased (Guangdong Guanghua) and used without further purification. IR spectra were obtained from KBr pellets on a Bruker EQUINOX 55 FT-IR spectrometer in the 400–4000 cm−1 region. Elemental analyses (C, H, N) were performed with a Vario EL elemental analyzer. Powder X-ray diffraction patterns were recorded by a Bruker D8 ADVANCE X-ray powder diffractometer (Cu-Kα). Thermogravimetric analyses were performed using a Netzsch STA 449C instrument with a heating rate of 5.0 °C min−1 under nitrogen atmosphere.Synthesis
Single crystals of the title compound were prepared by heating a mixture of Zn(NO3)2·6H2O (0.060 g, 0.2 mmol), Hip (0.044 g, 0.2 mmol), triethylamine (0.1 mL) and EtOH (or MeOH) (8.0 mL) in a sealed 12-mL Teflon-lined reactor at 180 °C for 3 days and then cooling at 5 °C h−1 to room temperature. A pure sample of the microcrystalline powder was prepared by heating a mixture of Zn(OH)2 (0.173 g, 1.75 mmol), Hip (0.660 g, 3 mmol), EtOH (40 mL) and aqueous ammonia (25%, 2.0 mL) sealed in a 50-mL Teflon-lined reactor, under stirring at 140 °C for 5 days. The light yellow resultant precipitate was isolated by filtration (yield 71%). Elemental analysis: Found: C 52.76, H 3.84, N 18.84. Calc.: C 52.44, H 3.84, N 18.82% for [Zn7(ip)12](OH)2·25H2O). IR: 3444vs, 1649m, 1456s, 1506m, 1425m, 1124w, 1080vw, 876w, 616w, 514vw cm−1. A thin film was prepared by immersing a zinc plate 1.5 × 1.5 cm2) in a solution of Hip (0.044 g, 0.2 mmol) in mixed NH3·H2O (25%, 0.1 mL) and EtOH (8.0 mL), which was sealed in a 12-mL Teflon-lined reactor, heated at 180 °C for 3 days and cooled to room temperature at a rate of 5 °C h−1.Single-crystal X-ray diffraction study
The intensity data were collected on a Bruker Apex CCD area-detector diffractometer (Mo-Kα). Absorption corrections were applied by using the multi-scan program SADABS. The structure was solved with the direct method and refined with a full-matrix least-squares technique with the SHELXTL program package. The guest molecules were refined as individual oxygen atoms. The organic hydrogen atoms were generated geometrically and refined in a riding model. No electron residue peak can be found nearby the uncoordinated nitrogen. Anisotropic thermal parameters were applied to all non-hydrogen atoms except the guest molecules.Gas sorption measurement
The sorption isotherms of N2 and CO2 were measured with automatic volumetric sorption apparatus (Belsorp MAX and HP). Before gas sorption measurements, the as-synthesized sample of 1·g was degassed under high vacuum at 100 °C for 12 h to entirely remove the remnant solvent molecules.Photoluminescent measurement
Steady-state photoluminescence spectra and lifetime measurements were performed on an Edinburgh FLS920 spectrometer (standard deviation 0.2 nm for λ) equipped with a continuous Xe900 xenon lamp and an nF900 nanosecond flash lamp. Except when using a 10−6 M solution of Hip, microcrystalline samples were used for all photoluminescence measurements. All instrument parameters such as excitation/emission slit and scanning speed were fixed for all measurements. Nevertheless, the luminescence intensity differences among the solvent-loaded samples are not accurate. On the other hand, luminescence intensity differences among 1·g, 1′, 1′·CO2 and 1·MeOH should be reliable, because the same powder sample was used for all measurements (in situ degas and introduction of CO2 in a sealed chamber equipped with quartz windows and a three-way valve which connects the chamber to a vacuum pump and a CO2 cylinder).Results and discussion
Synthesis and structure analysis
Single crystals of [Zn7(ip)12](OH)2·guest (1·g, MAF-34) suitable for X-ray diffraction study were prepared by solvothermal reaction of Zn(NO3)2/Zn(ClO4)2, Hip and triethylamine in EtOH or MeOH. Microcrystalline powder of 1·g was synthesized in high yield by replacing the soluble Zn(II) salt and base by Zn(OH)2, which was used in subsequent property studies.Single-crystal X-ray analysis‡ revealed that 1·g crystallizes in the cubic space group I![[4 with combining macron]](https://www.rsc.org/images/entities/char_0034_0304.gif) 3d, containing two independent Zn centres (Zn1 at a C3 axis, Zn2 at an S4 axis), and a deprotonated ip− ligand, as well as some disordered counter anions and guest molecules in the asymmetric unit. Zn1 is chelated by three phenanthroline moieties to furnish a distorted octahedral geometry (Fig. 1a). Zn2 is coordinated by four imidazolate moieties in a tetrahedral geometry (Fig. 1b). Each ip− ligand uses only three N atoms to coordinate two different Zn atoms, leaving an imidazolate N atom deprotonated and uncoordinated. Coordination of the imidazole N atom of Hip has been only reported in two recent examples, in which the imidazole moieties remain protonated.7,9 In other words, 1·g is the first coordination polymer using anionic ip− as a bridging ligand. The unsymmetric coordination of ip− obviously distorts the Zn(ip)3 moiety from the regular D3 symmetry observed in common M(L)3 structures (L = 2,2′-bipyridine-like chelating ligands). The degree of such local distortion may show a response to external stimuli.
3d, containing two independent Zn centres (Zn1 at a C3 axis, Zn2 at an S4 axis), and a deprotonated ip− ligand, as well as some disordered counter anions and guest molecules in the asymmetric unit. Zn1 is chelated by three phenanthroline moieties to furnish a distorted octahedral geometry (Fig. 1a). Zn2 is coordinated by four imidazolate moieties in a tetrahedral geometry (Fig. 1b). Each ip− ligand uses only three N atoms to coordinate two different Zn atoms, leaving an imidazolate N atom deprotonated and uncoordinated. Coordination of the imidazole N atom of Hip has been only reported in two recent examples, in which the imidazole moieties remain protonated.7,9 In other words, 1·g is the first coordination polymer using anionic ip− as a bridging ligand. The unsymmetric coordination of ip− obviously distorts the Zn(ip)3 moiety from the regular D3 symmetry observed in common M(L)3 structures (L = 2,2′-bipyridine-like chelating ligands). The degree of such local distortion may show a response to external stimuli.
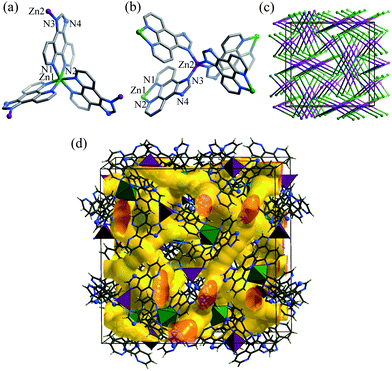 | ||
| Fig. 1 (a) Coordination environment of the distorted octahedral (Zn1–N1 2.172(5), Zn1–N2 2.133(5) Å; N1–Zn1–N2 77.0(2), N1–Zn–N1* 89.8(2), N2–Zn1–N2* 97.2(2)°) and (b) tetrahedral (Zn2–N3 1.98(5); N3–Zn–N3* 107.3(2) and 113.9(2)°) zinc atoms, (c) simplified framework topology (Zn1: green sphere, Zn2: purple sphere, ip−: stick), and (d) channel structure of 1·g. | ||
Deprotonation of the uncoordinated imidazole nitrogen atom in 1·g is very important because it determines the number of counter anions and also pore surface property. Although X-ray diffraction cannot reliably confirm the existence or otherwise of a proton, we have obtained enough evidences to prove the deprotonation of the imidazole moiety. Zn(OH)2 was adopted as the zinc ion source to make sure that anions can be only the conjugate base of the solvent. Although the acidity of imidazole is weak (pKa = 14.52), increasing the size of conjugate system on the imidazole ring (for 1H-benzimidazole, pKa = 12.78),10 adding an electron-withdrawing atom (for 1,2,4-triazole, pKa = 10.3) or coordinating with metal ions would all strengthen its acidity. Therefore, Hip (pKa = 8.08 calculated by ACD/Labs V11.02) in 1·g should have stronger acidity than the solvents used (pKa > 15), and the deprotonated species should be the imidazole moieties rather than the guest molecules. Nevertheless, complete deprotonation still can not fulfill charge balance requirement, and the two additional counter anions are assigned as hydroxide, because in the reaction system the strongest acid except for Hip (too large to be included in the small channels as counter anions) is H2O.11
Regarding Zn1 as a 3-connected node, Zn2 as a 4-connected node, and ip− as a linker, the whole structure extends into a distorted (3,4)-connected C3N4-type topology (ctn) (Fig. 1c), which has been only observed in a few coordination polymers.12 Despite the large volume of ip−, the long distance between the nodes (Zn1⋯Zn2 8.07 Å) results in an open framework (void = 47.6%) with complicated three-dimensional intersecting channels composed of small cavities (d = 4–6 Å) and apertures (d = 3.3 Å), which are occupied by OH− anions and guest molecules (Fig. 1d).
Framework stability and flexibility
Thermogravimetric analysis of 1·g showed complete removal of the guest molecules below 140 °C, and no obvious weight loss below 600 °C, which is much higher than for most PCPs (Fig. S1, ESI†).13 However, variable-temperature powder X-ray diffraction (PXRD) measurements revealed that the diffraction peaks become very weak and broad even at 40 °C (Fig. S2, ESI†), indicating the formation of a quasi-amorphous phase (1′).The completely activated sample 1′ can be obtained by heating 1·g at 140 °C at ambient pressure for 10 min, or heating at 100 °C at reduced pressure for 10 min, or by degassing 1·g at room-temperature for 1 h. PXRD showed that 1′ can transform back to phase 1·g with high crystallinity by adsorption of MeOH or EtOH, instead of H2O, benzene (BEN), or nitrobenzene (NBEN) (Fig. S3 and S4, Table S1, ESI†). Nevertheless, 1′ does not decompose in H2O, which reflects the strong Zn–ip coordination bonds. Interesting phase transformation phenomena was observed when 1·g was immersed in MeOH or EtOH at room temperature. It lost crystallinity and initially changed to a quasi-amorphous phase similar to 1′, but changed back to the original phase 1·g by keeping the sample in the same solvents for another few days (Fig. 2).14 These crystal-to-amorphous-to-crystal structural transformation processes during desolvation or guest exchange indicate that the framework can only keep its long-range order when it is completely filled with a single kind of guest molecules. Without guest or when the guest molecules are disordered, the framework becomes amorphous. The reversibility of these transformations suggests that the structural origin of amorphization should arise from distortion of the framework rather than bond cleavage.
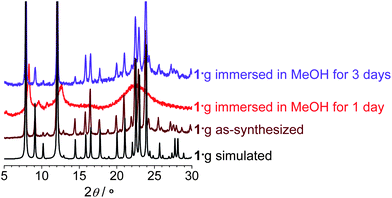 | ||
| Fig. 2 PXRD patterns of 1·g treated with different conditions. | ||
Gas sorption
To examine the porosity of 1′ toward gas molecules, CO2 and N2 sorption experiments have been performed. 1′ does not adsorb N2 at 77 K, which may be ascribed to framework contraction or diffusion problems at extremely low temperature, while it can adsorb considerable amounts of CO2 at 195 K. Room-temperature sorption isotherms were also measured up to 42 atm. The sorption amounts, for both CO2 and N2, are higher than those at low temperatures, although the room temperature measurements have much lower P/P0 (Fig. 3). Moreover, the CO2 isotherms revealed two sequential processes. At low pressures, the adsorption represents typical type-I isotherms and reaches ca. 20 cm3 g−1, indicating there are still small amounts of micropores in the quasi-amorphous 1′. Adsorption enthalpies of CO2 were calculated by the Clausius–Clapeyron equation and also fitted by the virial equation from adsorption isotherms measured at 273 and 298 K (Fig. 3b, S5 and S6, ESI†). The CO2 adsorption enthalpy at zero coverage 38.1 kJ mol−1 is comparable to those of adsorbents functionalized with open metal sites and amino groups,15 which may be ascribed to the uncoordinated imidazolate nitrogen donors on the pore surface of 1′. After the micropores are filled, the framework expands to accommodate more incoming CO2 molecules, which is easier at higher temperature and pressure. Nevertheless, saturation has not been attained at 195 K and P/P0 = 1, or at room temperature and 42 atm. The more pronounced hysteresis at room temperature might be attributed to the higher degree of framework expansion and exposure of more uncoordinated imidazolate N donors. These phenomena are related to the presence of anions in the ultramicropores and framework distortion of the activated sample, which would both block the diffusion of guest molecules, especially at low temperature.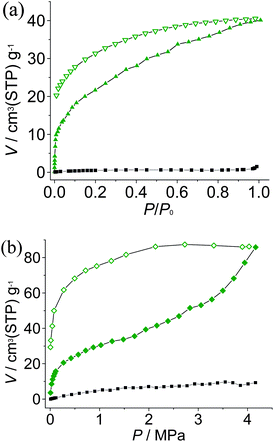 | ||
| Fig. 3 Sorption isotherms (solid and open symbols denote adsorption and desorption, respectively) of CO2 (green) and N2 (black) at (a) low temperature (195 K for CO2 and 77 K for N2) and (b) 298 K. | ||
Photoluminescent response
Photoluminescent properties of the microcrystalline samples were investigated at room temperature (Fig. 4, Fig. S7 and S8, ESI†). As-synthesized 1·g displays strong cyan photoluminescence (λmax = 487 nm). In contrast, the emission color of desolvated 1′ is orange (λmax = 554 nm). 1′ showed reversible luminescence responses toward different guests (solvent vapor or gas). Adsorption of MeOH/EtOH turns the emission color back to cyan (λmax = 480 nm). Notably, this exceptionally large shifting of emission λmax (74 nm) has been rarely documented for PCPs.6a, 16 On the other hand, BEN and H2O shift the emission color to yellow (λmax = 513 nm), which is also significantly blue-shifted from that of 1′. More dramatically, NBEN quenches the luminescence by more than 95%.17 Low-pressure CO2 also shifted the emission color of 1′ to yellowish orange (λmax = 540 nm). In situ photoluminescence measurements showed that the λmax and luminescent intensity are dependent on the pressure of CO2, which has not been reported previously for PCPs. These luminescent response processes could be repeated for at least three cycles, in which the guest-loaded samples can be readily activated again by heating at 140 °C for several minutes. Preliminary experiments showed that a luminescent thin film of 1·g can be directly grown on a metal zinc plate under solvothermal conditions (Fig. 4b and Fig. S9, ESI†), implying the potential for sensor applications.18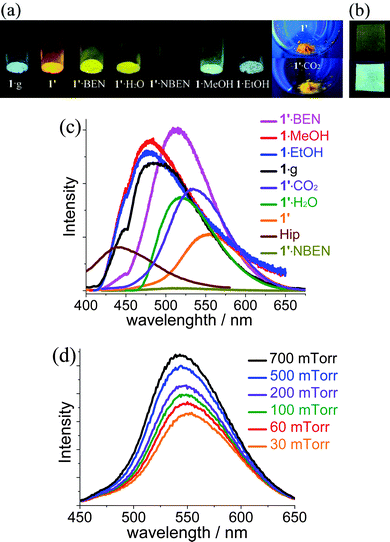 | ||
| Fig. 4 (a) Photographs of 1 in different guest inclusion states (365-nm UV light) and (b) 1·g thin film grown on a zinc plate (upper: ambient light, lower: 365-nm UV light). (c) Photoluminescence profiles of corresponding samples and (d) 1′ under variable pressure of CO2. | ||
Considering that adsorption of EtOH/MeOH can transform 1′ back to 1·g and the similar emission colors among 1·g, 1·EtOH and 1·MeOH, the luminescence response mechanism should arise from the guest-dependent network distortion. Although 1′ remains quasi-amorphous by treating with BEN/H2O/NBEN, the shifting of emission λmax and the quenching of luminescence could be ascribed to the host–guest interactions such as hydrogen bonds between the uncoordinated imidazolate N donors and H2O and π–π stacking interaction between the large aromatic ligand ip− and the aromatic guest molecules, so that the energy level of the ground state and excited state were changed. Especially, electrons may transfer to the electron-deficient NBEN in the excited state. The mechanism of hypsochromic shift response to CO2 should be similar with that for H2O. Nevertheless, for CO2 we can easily control the gas pressure at very low P/P0 and observe a small hypsochromic effect (Fig. 4d).
To further explore the luminescent mechanisms, the photoluminescence lifetimes of related samples were measured (Fig. S10 and Table S2, ESI†). Solid Hip exhibits no obviously detectable photoluminescence at room temperature, which may be due to the extensive π–π interactions of the large planar molecules and/or hydrogen bonding of the imidazole moities, facilitating energy transfer from one excited Hip to another Hip. A dilute Hip solution (10−6 M) in methanol shows blue emission (λmax = 440 nm). The emission decays of 1·g, 1·EtOH, 1·MeOH contain two components in the nanosecond range, being very similar to that of Hip. Therefore, the luminescence of these samples should be originated mainly from metal-perturbed ligand-centered transitions. On the other hand, the emission decay curves of 1′, 1′·BEN, 1′·H2O and 1′·CO2 are similar and must be fitted by a triple exponential function.19 Nevertheless, based on nanosecond lifetimes, all sample emissions are singlet to singlet, and they should be fluorescent in nature. Compared to the highly crystalline phases, the red-shift of emission color and additional emission component in the quasi-amorphous phases might be caused by ligand–ligand interaction in the distorted framework.20 Detailed examination showed that adjacent ip− ligands in 1·g are separated by the Zn ions to avoid typical face-to-face or edge-to-face π–π interactions. However, they are still located very close to each other, and may be even closer when the framework is distorted in the quasi-amorphous phase (Fig. S11, ESI†).
Conclusions
In summary, an old ligand was successfully utilized in a new way for the construction of a flexible, luminescent, and porous framework with not only unique network topology but also active uncoordinated imidazolate nitrogen donors on the pore surface. The drastic structural and photoluminescence responses toward a variety of guest molecules indicate that the material may be used for multi-purpose sensing applications.Acknowledgements
This work was supported by the “973 Program” (2007CB815302 & 2012CB821706), NSFC (20821001 & 21001120), Chinese Ministry of Education (109125 & ROCS), and the Open Fund of the State Key Laboratory of Optoelectronic Materials and Technologies.Notes and references
- (a) M. D. Allendorf, C. A. Bauer, R. K. Bhakta and R. J. T. Houk, Chem. Soc. Rev., 2009, 38, 1330–1352 RSC; (b) G. Férey and C. Serre, Chem. Soc. Rev., 2009, 38, 1380–1399 RSC; (c) S. Horike, S. Shimomura and S. Kitagawa, Nat. Chem., 2009, 1, 695–704 CrossRef CAS.
- (a) Y. Li, G. Xu, W.-Q. Zou, M.-S. Wang, F.-K. Zheng, M.-F. Wu, H.-Y. Zeng, G.-C. Guo and J.-S. Huang, Inorg. Chem., 2008, 47, 7945–7947 CrossRef CAS; (b) B. L. Chen, L. B. Wang, Y. Q. Xiao, F. R. Fronczek, M. Xue, Y. J. Cui and G. D. Qian, Angew. Chem., Int. Ed., 2009, 48, 500–503 CrossRef CAS; (c) Y.-H. Wang, B. Li, Y.-H. Liu, L.-M. Zhang, Q.-H. Zuo, L.-F. Shi and Z.-M. Su, Chem. Commun., 2009, 5868–5870 RSC; (d) A. Lan, K. Li, H. Wu, D. H. Olson, T. J. Emge, W. Ki, M. Hong and J. Li, Angew. Chem., Int. Ed., 2009, 2334–2338 CAS; (e) Z. G. Xie, L. Q. Ma, K. E. deKrafft, A. Jin and W. B. Lin, J. Am. Chem. Soc., 2010, 132, 922–923 CrossRef CAS; (f) D. Yuan, D. Zhao, D. J. Timmons and H.-C. Zhou, Chem. Sci., 2011, 2, 103–106 RSC; (g) C. Wang and W. B. Lin, J. Am. Chem. Soc., 2011, 133, 4232–4235 CrossRef CAS.
- J. P. Zhang and X. M. Chen, Chem. Commun., 2006, 1689–1699 RSC.
- (a) Y. Q. Tian, C. X. Cai, Y. Ji, X. Z. You, S. M. Peng and G. H. Lee, Angew. Chem., Int. Ed., 2002, 41, 1384–1386 CrossRef CAS; (b) X. C. Huang, J. P. Zhang and X. M. Chen, Chin. Sci. Bull., 2003, 48, 1531–1534 CAS; (c) X. C. Huang, Y. Y. Lin, J. P. Zhang and X. M. Chen, Angew. Chem., Int. Ed., 2006, 45, 1557–1559 CrossRef CAS; (d) R. Banerjee, A. Phan, B. Wang, C. Knobler, H. Furukawa, M. O'Keeffe and O. M. Yaghi, Science, 2008, 319, 939–943 CrossRef CAS.
- (a) S. A. Moggach, T. D. Bennett and A. K. Cheetham, Angew. Chem., Int. Ed., 2009, 48, 7087–7089 CrossRef CAS; (b) C. Gücüyener, J. van den Bergh, J. Gascon and F. Kapteijn, J. Am. Chem. Soc., 2010, 132, 17704–17706 CrossRef; (c) S. Aguado, G. Bergeret, M. P. Titus, V. Moizan, C. Nieto-Draghi, N. Bats and D. Farrusseng, New J. Chem., 2011, 35, 546–550 RSC.
- (a) D. Tanaka, S. Horike, S. Kitagawa, M. Ohba, M. Hasegawa, Y. Ozawa and K. Toriumi, Chem. Commun., 2007, 3142–3144 RSC; (b) K. C. Stylianou, R. Heck, S. Y. Chong, J. Bacsa, J. T. A. Jones, Y. Z. Khimyak, D. Bradshaw and M. J. Rosseinsky, J. Am. Chem. Soc., 2010, 132, 4119–4130 CrossRef CAS.
- S. Shi, J. Liu, T. M. Yao, X. T. Geng, L. F. Jiang, Q. Y. Yang, L. Cheng and L. N. Ji, Inorg. Chem., 2008, 47, 2910–2912 CrossRef CAS.
- (a) W. Paw and R. Eisenberg, Inorg. Chem., 1997, 36, 2287–2293 CrossRef CAS; (b) B. Jing, T. Wu, C. Tian, M. Zhang and T. Shen, Bull. Chem. Soc. Jpn., 2000, 73, 1749–1755 CrossRef CAS.
- J.-Q. Liu, Y.-N. Zhang, Y.-Y. Wang, J.-C. Jin, E. K. Lermontova and Q.-Z. Shi, Dalton Trans., 2009, 5365–5378 RSC.
- R. W. I. Harold Walba, J. Org. Chem., 1961, 26, 2789–1791 CrossRef.
- (a) Q.-G. Zhai, C.-Z. Lu, S.-M. Chen, X.-J. Xu and W.-B. Yang, Cryst. Growth Des., 2006, 6, 1393–1398 CrossRef CAS; (b) H. A. Habib, J. Sanchiz and C. Janiak, Dalton Trans., 2008, 1734–1744 RSC; (c) Z. Li, M. Li, S.-Z. Zhan, X.-C. Huang, S. W. Ng and D. Li, CrystEngComm, 2008, 10, 978–980 RSC.
- (a) D. N. Dybtsev, H. Chun and K. Kim, Chem. Commun., 2004, 1594–1595 RSC; (b) Z. R. Pan, X. Q. Yao, H. G. Zheng, Y. Z. Li, Z. J. Guo and S. R. Batten, CrystEngComm, 2009, 11, 605–609 RSC; (c) S. M. Chen, J. Zhang, T. Wu, P. Y. Feng and X. H. Bu, J. Am. Chem. Soc., 2009, 131, 16027–16029 CrossRef CAS.
- M. J. Rosseinsky, Microporous Mesoporous Mater., 2004, 73, 15–30 CrossRef CAS.
- C.-D. Wu and W. Lin, Angew. Chem., Int. Ed., 2005, 44, 1958–1961 CrossRef CAS.
- (a) D. M. D'Alessandro, B. Smit and J. R. Long, Angew. Chem., Int. Ed., 2010, 49, 6058–6082 CrossRef CAS; (b) R. Vaidhyanathan, S. S. Iremonger, K. W. Dawson and G. K. H. Shimizu, Chem. Commun., 2009, 5230–5232 RSC.
- (a) E. Y. Lee, S. Y. Jang and M. P. Suh, J. Am. Chem. Soc., 2005, 127, 6374–6381 CrossRef CAS; (b) Y.-Q. Huang, B. Ding, H.-B. Song, B. Zhao, P. Ren, P. Cheng, H.-G. Wang, D.-Z. Liao and S.-P. Yan, Chem. Commun., 2006, 4906–4908 RSC; (c) R. Grünker, I. Senkovska, R. Biedermann, N. Klein, A. Klausch, I. A. Baburin, U. Mueller and S. Kaskel, Eur. J. Inorg. Chem., 2010, 3835–3841 CrossRef; (d) A. Kobayashi, H. Hara, S.-i. Noro and M. Kato, Dalton Trans., 2010, 39, 3400–3406 RSC; (e) Y. Takashima, V. M. Martinez, S. Furukawa, M. Kondo, S. Shimomura, H. Uehara, M. Nakahama, K. Sugimoto and S. Kitagawa, Nat. Commun., 2011, 2, 168–175 CrossRef.
- (a) S. Pramanik, C. Zheng, X. Zhang, T. J. Emge and J. Li, J. Am. Chem. Soc., 2011, 133, 4153–4155 CrossRef CAS; (b) H. Sohn, M. J. Sailor, D. Magde and W. C. Trogler, J. Am. Chem. Soc., 2003, 125, 3821–3830 CrossRef CAS.
- (a) G. Lu and J. T. Hupp, J. Am. Chem. Soc., 2010, 132, 7832–7833 CrossRef CAS; (b) X. Q. Zou, G. S. Zhu, I. J. Hewitt, F. X. Sun and S. L. Qiu, Dalton Trans., 2009, 3009–3013 RSC.
- (a) C. A. Kent, B. P. Mehl, L. Ma, J. M. Papanikolas, T. J. Meyer and W. Lin, J. Am. Chem. Soc., 2010, 132, 12767–12769 CrossRef CAS; (b) C. A. Kent, D. Liu, L. Ma, J. M. Papanikolas, T. J. Meyer and W. Lin, J. Am. Chem. Soc., 2011 DOI:10.1021/ja204214t.
- C. A. Bauer, T. V. Timofeeva, T. B. Settersten, B. D. Patterson, V. H. Liu, B. A. Simmons and M. D. Allendorf, J. Am. Chem. Soc., 2007, 129, 7136–7144 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Additional structural plots, PXRD, TGA curves, additional sorption isotherms, luminescenct profiles, as well as X-ray crystallographic files in CIF format. CCDC reference number 826298. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c1sc00421b |
| ‡ Crystal data for 1·g: C156H84N48O68Zn7, M = 4176.30, T = 113(2) K, cubic, space group I3d, a = 27.170(2) Å, V = 20058(2) Å3, Z = 4, Dc = 1.383 g cm−3, R1 = 0.0615 (I > 2σ(I)), wR2 = 0.1596 (all data), S = 1.048.. |
| This journal is © The Royal Society of Chemistry 2011 |