Unusual rutileTiO2 nanosheets with exposed (001) facets†
Jun Song
Chen
and
Xiong Wen (David)
Lou
*
School of Chemical and Biomedical Engineering, Nanyang Technological University, 70 Nanyang Drive, Singapore, 637457, Singapore. E-mail: xwlou@ntu.edu.sg
First published on 18th August 2011
Abstract
In this work, we report an interesting synthesis of unusual rutileTiO2 nanosheets with exposed high-energy (001) facets, which form hierarchical microspheres that closely resemble the natural “desert rose”. These high-energy (001) surfaces are stabilized by amorphous molybdenum trioxide (MoO3), which is homogeneously dispersed on the TiO2 nanosheets. The as-prepared unique TiO2–MoO3 hybrid structure exhibits improved lithium storage capabilities.
Introduction
The engineering of nanocrystal facets has been gaining increasing research interest in materials science. Through exposing facets with a high surface energy, one is able to obtain unique nanostructured materials with superior bulk or surface properties for a myriad of applications.1–4 Recently, anatase titanium dioxide (TiO2) has been successfully synthesized in the form of nanosheets with an exposed (001) facet, which possesses the highest surface energy among the low-index facets of anatase TiO2. In the synthesis, fluorine was employed to effectively stabilize the (001) high-energy facet,5 thus preventing it from diminishing during the crystal growth process. As summarized in a recent review,6 numerous approaches have then been developed to achieve intriguing anatase TiO2 nanostructures with exposed (001) facets utilizing different precursors and surface stabilizing agents.7–15 Such anatase TiO2 crystals with exposed (001) facets have been found to manifest enhanced physicochemical properties in photocatalysis and lithium storage.6On the other hand, rutile TiO2, which is the most thermodynamically stable polymorph in the titania family,16 has been less studied. Analogous to the anatase form, rutile TiO2 also possesses a tetragonal crystal structure with the (001) plane possessing the highest surface energy of 28.9 meV a.u.−2 , and the (110) facet being the most stable one with a surface energy of only 15.6 meV a.u.−2.17 Even though many synthesis systems of rutile TiO2 have been proposed, the products are generally limited to nanoparticles16,18,19 or one-dimensional rod-like structures.20–25 Thus, it is intrinsically challenging to synthesize rutile TiO2 crystals with exposed (001) facets, which are anticipated to exhibit enhanced properties when compared to rutile crystals in the normal forms of nanorods20–25 or nanoparticles.16,18,19
To this end, herein, we report a facile hydrothermal synthesis of rutile TiO2 hierarchical microspheres assembled from nanosheets with exposed (001) facets, that closely resemble the natural “desert rose”.26 These high-energy surfaces are stabilized by amorphous molybdenum trioxide (MoO3), which is homogeneously dispersed on the TiO2 nanosheets. To our knowledge, this is the first example of the successful construction of rutileTiO2 nanosheets with exposed (001) high-energy facets stabilized by an amorphous metal oxide. Furthermore, the as-prepared TiO2-MoO3 hybrid structure exhibits enhanced lithium storage capabilities when compared to pure rutile nanoparticles, as well as other rutile TiO2 nanomaterials.
Results and discussion
In a typical synthesis, ammonium heptamolybdate tetrahydrate (AHM) and TiF4 solution were used as the precursors for MoO3 and TiO2, respectively. The reaction was carried out in an acidic environment at an elevated temperature for a relatively short duration. Fig. 1A shows the schematic drawing of the crystal structures of rutile TiO2: I is the Wulff construction model based on the surface energies of different facets at equilibrium, while II displays the structure of nanosheet with exposed (001) facets synthesized in the present study showing a square-shaped platelet structure with four truncated corners. Fig. 1B depicts a low-magnification scanning electron microscopy (SEM) image. It is clear that the as-prepared TiO2–MoO3 product consists of microspheres of approximately one micrometre in diameter, with a relatively uniform size distribution (also see the supporting information, Fig. S1†). A closer examination (Fig. 1C) reveals that these microspheres are composed of nanosheets forming a hierarchical structure with a great resemblance to the natural “desert rose” (Fig. 1B, inset). The surface features of these spheres are further revealed under a higher magnification (Fig. 1D), which consists of randomly-oriented sheet-like structures with a thickness in the range of 10–20 nm. Such a hierarchical structure possesses a specific surface area of 34 m2 g−1 (the N2 adsorption–desorption isotherm is provided in Fig. S2†).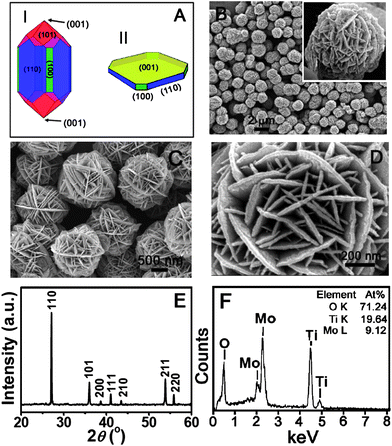 | ||
| Fig. 1 (A) A schematic drawing of the Wulff construction of a rutile TiO2 crystal at equilibrium (I) and the as-formed nanosheet with exposed (001) facet (II). (B–D) Field-emission scanning electron microscopy (FESEM) images. (D) X-ray diffraction (XRD) pattern and (E) Energy-dispersive X-ray (EDX) analysis of the as-prepared TiO2–MoO3 composite. The inset in (B) shows a natural desert rose. | ||
The x-ray diffraction (XRD) pattern of the as-prepared sample is shown in Fig. 1E. All of the identified peaks can be indexed to the tetragonal rutile-phase TiO2 (JCPDS No. 21-1276, S.G.: P42/mnm, ao = 4.5933 Å, co = 2.9592 Å). Remarkably, no peaks of molybdenum oxide phases are identified in the XRD pattern, while the presence of the element Mo is confirmed by the energy-dispersive X-ray (EDX) analysis (Fig. 1F). Thus, we hypothesize that molybdenum oxide exists as an amorphous phase in the hybrid structure.
Fig. 2A shows a transmission electron microscope (TEM) image of a single TiO2–MoO3 microsphere. It can be observed that the outer region of the sphere is composed of very thin nanosheets, while the inner core is more densely packed. The high-resolution (HR) TEM image (Fig. 2B) of a representative single nanosheet clearly shows a pair of visible lattice fringes that are perpendicular to each other. Interestingly, a number of amorphous domains are observable on the surface (highlighted by white dotted circles), leading to the observed discontinuity of the crystal lattice. The selected-area electron diffraction (SAED) pattern (Fig. 2B, inset) of the nanosheet depicts a spot pattern that can be unambiguously indexed to the [001] zone of rutile TiO2. The HRTEM image of the area marked with a black square in Fig. 2B is given in Fig. 2C and the two sets of lattices have an equal interplanar distance of 0.64 nm, which corresponds to the (110) plane of rutile TiO2. Therefore, it can be concluded that both the upper and lower surfaces of the nanosheets are bounded by the (001) facets.
 | ||
| Fig. 2 (A) A transmission electron microscopy (TEM) image of the as-prepared TiO2–MoO3 composite. (B) A TEM image of a single nanosheet with visible lattice fringes. The inset shows the selected-area electron diffraction (SAED) pattern of the nanosheet. (C)A high-resolution (HR) TEM image of the region marked by the black square in (B). | ||
The elemental composition of the sample was further studied by the local element mapping technique mounted to HRTEM, giving the result shown in Fig. 3. It is apparent that the particle has uniform distributions of all the elements (Mo, Ti, O), both at the central region of the sphere (Fig. 3A) and the peripheral part of the nanosheets (Fig. 3B). The X-ray photoelectron spectroscopy (XPS) analysis (Fig. S3†) proves that the element Mo is indeed in its highest oxidation state of +6, while the oxidation state of Ti is also consistent with Ti(IV) reported previously.27 Furthermore, the Laser Raman spectra (Fig. S4†) indicate no presence of crystalline α-MoO3 in the TiO2–MoO3 hybrid structure, which is consistent with the above XRD analysis. All of these observations support the earlier hypothesis that the nanosheets are rutile TiO2 with exposed (001) facets that are stabilized by amorphous MoO3. Indeed, several previous reports have proved that MoO3 has a high tendency to be dispersed onto the surface of other oxide materials, including SiO2, Al2O3 and TiO2,28–30 showing the strongest interactions with titania supports.30 It is thus conceivable that the amorphous MoO3 forms bonds with the highly reactive (001) surfaces of rutile TiO2, which provides the enhanced stabilities of these high-energy facets. The possibility of containing an appreciable amount of hydrate in the composite is ruled out by thermal gravimetric analysis (Figure S5). It shows a weight loss of only 6% up to 600 °C, which can be attributed to the evaporation of adsorbed moisture content.
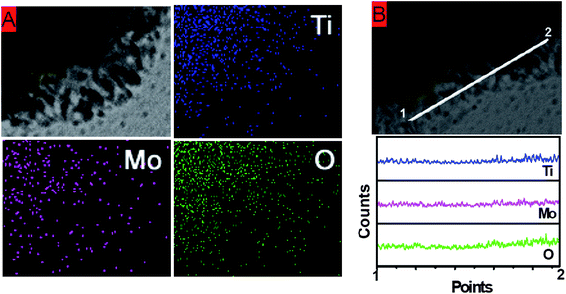 | ||
| Fig. 3 (A) Element mapping over part of the composite sphere. (B) Element mapping along a tangential line at the nanosheet region. | ||
In order to understand the formation of this unique structure, time-series experiments are performed (Fig. S6†). It shows that when the reaction duration is shortened to only 2 h, some platelet-like structures can be identified budding out from the central sphere-like cores. On the other hand, when the synthesis is prolonged to 12 h, the nanosheet constituents appear to be more pronounced. However, the overgrowth of these subunits leads to the aggregation among the microspheres. Other control experiments were also performed, in which only one of the metal oxide precursors (either AHM or TiF4) is present in the reaction system while other conditions were kept unchanged. Noticeably, large platelet-like structures without defined shapes were formed from AHM, whereas the reaction with a sole TiF4 precursor will produce interconnected anatase TiO2 structures consisting of square-shaped subunits with a very low product yield. This suggests that the desired structure can only be constructed when both components are present and the homogeneous dispersion of amorphous MoO3 on the (001) facets of rutile TiO2 plays a crucial role in the formation of the “desert rose”-like structure. It is also important to mention that even slight variations in the concentrations of these two precursors or HNO3 acid (Table S1†) will result in products with distinct morphologies (Fig. S7†).
Rutile TiO2 has been intensively investigated for application in electrochemical lithium storage.16,23,31 However, its performance is hampered by the highly anisotropic diffusion of Li+ ions in the rutile structure. The diffusion coefficient of Li+ ions along the c-direction is ca. 10−6 cm2s−1 and this becomes 9 orders smaller in the ab-plane.32 Consequently, it is suggested that Li+ insertion into micrometre-sized rutile particles is kinetically restricted at room temperature32 and full Li loading can only be achieved at a minimum temperature of 120 °C.33 It has been recently reported that rutileTiO2 nanoparticles can exhibit enhanced lithium storage capabilities.16,23 Nevertheless, for rutile TiO2 crystals normally dominated by the most stable (110) surfaces, there are hardly any open channels available for Li+ ions to enter the bulk and so the insertion is largely a surface effect,32 which leads to very limited lithium storage capabilities. Since the as-prepared TiO2–MoO3 hybrid structure in the present study possesses largely exposed reactive (001) facets on the surface, more channels along the c-direction are available to facilitate Li+ ion insertion/extraction. Therefore one would expect an enhanced lithium storage capability for this unique hybrid structure. To verify this idea, its electrochemical properties were investigated. Fig. 4A displays cyclic voltammograms (CV) of the as-prepared sample. Generally, it shows a similar featureless pattern to those reported previously.19 Only in the first cathodic scan, two broad peaks appear at about 1.6 V and 2.25 V, which quickly become indiscernible in the subsequent cycles. The charge–discharge profile of the hybrid material at 50 mA g−1 is shown in Fig. 4B, which is generally consistent with above CV behavior and previous reports.31 There are two poorly defined plateau regions at about 2.3 V and 1.8 V during the first discharge, leading to a high specific capacity of 379 mA h g−1. A reversible charge capacity of 246 mA h g−1 can be obtained, giving rise to a capacity loss of about 35%. This relatively large initial loss of capacity is attributed to the trapping of initially inserted Li+ ions in the rutile structure, which makes full extraction hard to achieve.32 The second discharge conveys a lower capacity of 278 mA h g−1 and the reversible charge capacity is 261 mA h g−1, resulting in a much lower capacity loss of only 6%.
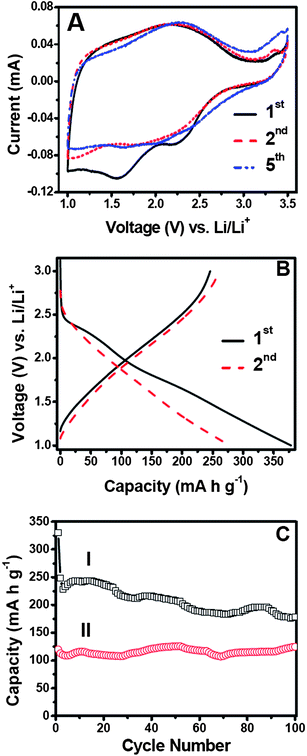 | ||
| Fig. 4 Electrochemical measurements of the as-prepared TiO2–MoO3 composite. (A) Representative cyclic voltammograms (CV) at a scan rate of 0.1 mV s−1 between 1–3.5 V. (B) The charge–discharge profile of the first (I) and second (II) cycles at a current rate of 50 mA g−1. (C) The cycling performance at a current rate of 600 mA g−1 (I). The performance of pure rutileTiO2 nanoparticles (II) under the same conditions is provided as a comparison. All of the charge–discharge tests were performed with a voltage window of 1–3 V. | ||
The cycling performance of the TiO2–MoO3 nanocomposite at a higher current rate of 600 mA g−1 is depicted in Fig. 4C and the results obtained at other current rates are also provided in Fig. S8†. It can be observed that a significant loss occurs after the first discharge capacity of 330 mA h g−1. The sample manifests good cyclic capacity retention upon extended cycling with a reversible capacity of 179 mA h g−1 at the end of 100 cycles. The average irreversible capacity loss can thus be calculated to be 0.46% per cycle. When compared to pure rutile TiO2 microparticles, the as-prepared TiO2–MoO3 nanocomposite exhibits a much higher capacity throughout the cycling. The rate capability behavior is also advantageous over the performance obtained from nanosized rod-like rutile TiO2,23 or even graphene-supported rutileTiO2 nanorods.31 The above evidence clearly proves that the unique rutileTiO2 nanosheets with exposed (001) high energy facets possess greatly enhanced electrochemical properties.
Conclusions
In summary, we have developed a facile hydrothermal method to synthesize a unique TiO2–MoO3 hybrid structure assembled from rutileTiO2 nanosheets with exposed (001) facets. Various characterizations prove that the uniform dispersion of amorphous MoO3 on the high-energy (001) facets of rutile TiO2 provides sufficient stability to preserve these surfaces, while the nature of the interaction between MoO3 and TiO2 requires more in-depth investigations. Furthermore, this hybrid structure exhibits improved lithium storage capabilities presumably due to the presence of a large number of (001) facets. By forming MoO3-stablized rutileTiO2 nanosheets with exposed (001) high-energy facets, this work presents a new concept of creating novel metal oxide hybrid structures with enhanced properties.Experimental section
Materials preparation
The as-formed TiO2–MoO3 composite microspheres with nanosheets were synthesized by a straightforward hydrothermal method. Hydrochloric acid was used to adjust the pH of the ultra pure water to about 2. A certain amount of titanium tetrafluoride (TiF4, Aldrich) was dissolved in this acidic solution to achieve a concentration of 0.04 M. Typically, 0.1 g of ammonium heptamolybdate tetrahydrate (AHM; (NH4)6Mo7O24·4H2O, Sigma-Aldrich) was dissolved in 42 ml of ultra pure water, followed by the addition of 2 ml of 65% HNO3 and 6 ml of the above acid-stabilized TiF4 solution. After shaking gently by hand, the mixture was transferred into a 60 ml Teflon-lined stainless steel autoclave, which was then kept in an electric oven at 180 °C for 5 h. After that, it was taken out to cool down to room temperature. The green–yellow colored precipitate was then collected by centrifugation and thoroughly washed with ultra pure water before drying at 60 °C overnight.Materials characterization
The crystalline structure of the product was confirmed by X-ray powder diffraction (XRD; Bruker, D8 - Advance X-Ray Diffractometer, Cu-Kα, λ = 1.5406 Å). The morphology, as well as the elemental composition, was revealed by field-emission scanning electron microscopy (FESEM; JEOL, JSM-6700F, 5 kV, with EDX) and field-emission transmission electron microscopy (FETEM; JEOL, JEM-2100F, 200 kV, with ED). The thermal behavior of the sample was analyzed by thermogravimetry (TG; Shimadzu, DRG-60). The chemical composition was investigated with X-ray photoelectron spectroscopy (XPS; PHI 5600). All of the binding energy (BE) peaks were referenced to the C 1s peak (BE = 284.9 eV).Electrochemical analysis
The electrochemical tests were performed using two-electrode Swagelok-type cells with lithium serving as both the counter and reference electrodes under ambient temperature. The working electrode was composed of 70 wt % of active material (e.g., TiO2–MoO3 composite), 20 wt % of conductivity agent (carbon black, Super P Li), and 10 wt % of binder (polyvinylidene difluoride, PVDF, Aldrich). The electrolyte used was 1 M LiPF6 in a 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50 (w/w) mixture of ethylene carbonate and diethyl carbonate. Cell assembly was carried out in an Argon-filled glovebox with both moisture and oxygen contents below 1 ppm. Cyclic voltammetry (CV, 1 V–3 V, 0.1 mV s−1) was performed using an electrochemical workstation (CHI 660C). Galvanostatic charge/discharge was performed using a battery tester (NEWAER).
:50 (w/w) mixture of ethylene carbonate and diethyl carbonate. Cell assembly was carried out in an Argon-filled glovebox with both moisture and oxygen contents below 1 ppm. Cyclic voltammetry (CV, 1 V–3 V, 0.1 mV s−1) was performed using an electrochemical workstation (CHI 660C). Galvanostatic charge/discharge was performed using a battery tester (NEWAER).
Acknowledgements
We gratefully appreciate financial support from Nanyang Technological University and the Ministry of Education (Singapore) through the AcRF Tier-1 grant (RG 63/08, M52120096).Reference
- N. Tian, Z. Y. Zhou, S. G. Sun, Y. Ding and Z. L. Wang, Science, 2007, 316, 732 CrossRef CAS.
- M. Schrinner, M. Ballauff, Y. Talmon, Y. Kauffmann, J. Thun, M. Moller and J. Breu, Science, 2009, 323, 617 CrossRef CAS.
- B. Lim, M. J. Jiang, P. H. C. Camargo, E. C. Cho, J. Tao, X. M. Lu, Y. M. Zhu and Y. N. Xia, Science, 2009, 324, 1302 CrossRef CAS.
- L. Xiao, L. Zhuang, Y. Liu, J. T. Lu and H. D. Abruna, J. Am. Chem. Soc., 2009, 131, 602 CrossRef CAS.
- H. G. Yang, C. H. Sun, S. Z. Qiao, J. Zou, G. Liu, S. C. Smith, H. M. Cheng and G. Q. Lu, Nature, 2008, 453, 638 CrossRef CAS.
- J. S. Chen, L. A. Archer and X. W. Lou, J. Mater. Chem., 2011, 21, 9912 RSC.
- J. S. Chen, D. Luan, C. M. Li, F. Y. C. Boey, S. Qiao and X. W. Lou, Chem. Commun., 2010, 46, 8252 RSC.
- J. S. Chen, Y. L. Tan, C. M. Li, Y. L. Cheah, D. Y. Luan, S. Madhavi, F. Y. C. Boey, L. A. Archer and X. W. Lou, J. Am. Chem. Soc., 2010, 132, 6124 CrossRef CAS.
- Y. Q. Dai, C. M. Cobley, J. Zeng, Y. M. Sun and Y. N. Xia, Nano Lett., 2009, 9, 2455 CrossRef CAS.
- X. Han, Q. Kuang, M. Jin, Z. Xie and L. Zheng, J. Am. Chem. Soc., 2009, 131, 3152 CrossRef CAS.
- B. H. Wu, C. Y. Guo, N. F. Zheng, Z. X. Xie and G. D. Stucky, J. Am. Chem. Soc., 2008, 130, 17563 CrossRef CAS.
- H. G. Yang, G. Liu, S. Z. Qiao, C. H. Sun, Y. G. Jin, S. C. Smith, J. Zou, H. M. Cheng and G. Q. Lu, J. Am. Chem. Soc., 2009, 131, 4078 CrossRef CAS.
- Y. W. Jun, M. F. Casula, J. H. Sim, S. Y. Kim, J. Cheon and A. P. Alivisatos, J. Am. Chem. Soc., 2003, 125, 15981 CrossRef CAS.
- M. Liu, L. Y. Piao, L. Zhao, S. T. Ju, Z. J. Yan, T. He, C. L. Zhou and W. J. Wang, Chem. Commun., 2010, 46, 1664 RSC.
- A. Selloni, Nat. Mater., 2008, 7, 613 CrossRef CAS.
- J. S. Chen and X. W. Lou, J. Power Sources, 2010, 195, 2905 CrossRef CAS.
- M. Ramamoorthy, D. Vanderbilt and R. D. Kingsmith, Phys. Rev. B, 1994, 49, 16721 CrossRef CAS.
- L. Kavan, D. Fattakhova and P. Krtil, J. Electrochem. Soc., 1999, 146, 1375 CrossRef CAS.
- N. A. Milne, M. Skyllas-Kazacos and V. Luca, J. Phys. Chem. C, 2009, 113, 12983 CAS.
- E. Baudrin, S. Cassaignon, M. Koesch, J. P. Jolivet, L. Dupont and J. M. Tarascon, Electrochem. Commun., 2007, 9, 337 CrossRef CAS.
- W. J. H. Borghols, M. Wagemaker, U. Lafont, E. M. Kelder and F. M. Mulder, Chem. Mater., 2008, 20, 2949 CrossRef CAS.
- X. J. Feng, J. Zhai and L. Jiang, Angew. Chem., Int. Ed., 2005, 44, 5115 CrossRef CAS.
- Y. S. Hu, L. Kienle, Y. G. Guo and J. Maier, Adv. Mater., 2006, 18, 1421 CrossRef CAS.
- X. Li, Y. Xiong, Z. Li and Y. Xie, Inorg. Chem., 2006, 45, 3493 CrossRef CAS.
- M. Vijayakumar, S. Kerisit, C. Wang, Z. Nie, K. M. Rosso, Z. Yang, G. Graff, J. Liu and J. Hu, J. Phys. Chem. C, 2009, 113, 14567 CAS.
- H. Chen and C. P. Grey, Adv. Mater., 2008, 20, 2206 CrossRef CAS.
- X. W. Lou and H. C. Zeng, J. Am. Chem. Soc., 2003, 125, 2697 CrossRef CAS.
- S. H. Elder, F. M. Cot, Y. Su, S. M. Heald, A. M. Tyryshkin, M. K. Bowman, Y. Gao, A. G. Joly, M. L. Balmer, A. C. Kolwaite, K. A. Magrini and D. M. Blake, J. Am. Chem. Soc., 2000, 122, 5138 CrossRef CAS.
- M. Delarco, S. R. G. Carrazan, C. Martin, V. Rives, J. V. Garciaramos and P. Carmona, Spectrochim. Acta, Part A, 1994, 50, 2215 CrossRef.
- A. N. Desikan, L. Huang and S. T. Oyama, J. Chem. Soc., Faraday Trans., 1992, 88, 3357 RSC.
- D. H. Wang, D. W. Choi, J. Li, Z. G. Yang, Z. M. Nie, R. Kou, D. H. Hu, C. M. Wang, L. V. Saraf, J. G. Zhang, I. A. Aksay and J. Liu, ACS Nano, 2009, 3, 907 CrossRef CAS.
- D. Deng, M. G. Kim, J. Y. Lee and J. Cho, Energy Environ. Sci., 2009, 2, 818 CAS.
- M. V. Koudriachova, N. M. Harrison and S. W. de Leeuw, Solid State Ionics, 2003, 157, 35 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, more SEM images, N2 adsorption–desoprtion isotherm, XPS data, Raman spectra, TGA result and electrochemical results. See DOI: 10.1039/c1sc00307k |
| This journal is © The Royal Society of Chemistry 2011 |