Total synthesis of hyacinthacine A2: stereocontrolled 5-aza-cyclooctene photoisomerization and transannular hydroamination with planar-to-point chirality transfer†
Maksim
Royzen
,
Michael T.
Taylor
,
Andrew
DeAngelis
and
Joseph M.
Fox
*
Brown Laboratories, Department of Chemistry and Biochemistry, University of Delaware, Newark, DE 19716, USA. E-mail: jmfox@udel.edu; Fax: (+1)-302-831-6335
First published on 18th August 2011
Abstract
The total synthesis of hyacinthacine A2 is reported via a novel transannular hydroamination in which planar chirality of a 5-aza-trans-cyclooctene precursor is transferred to point chirality in the product. Key to the success of this strategy was the development of a method for establishing absolute planar chirality via stereocontrolled photoisomerization of a 5-aza-cis-cyclooctene. This was accomplished by constructing a 5-aza-cis-cyclooctene precursor with a trans-fused acetonide. The improved diastereoselectivity observed upon photoisomerization of this derivative is attributed to the conformational strain of the eight-membered ring in the minor diastereomer.
Introduction
The pyrrolizidine framework is common to manifold natural products,1 including the polyhydroxylated natural products hyacinthacine A2 (1), australine, alexine and their epimers (Fig. 1). These natural products and analogs have been targeted for synthesis1–5 because of their selective glycosidase inhibition6 and activity against HIV.7 For example, hyacinthacine A2 (8.6 μM),8 australine (5.8 μM),9 and alexine (11 μM)10 are all selective inhibitors of amyloglucosidase that do not display significant inhibition of β-glucosidases. Because of the diverse biological activities of pyrrolizidine alkaloids,1 the stereocontrolled construction of this privileged scaffold is of considerable interest.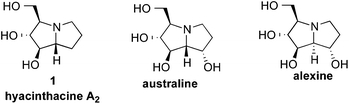 | ||
| Fig. 1 Representative pyrrolizidine alkaloid natural products. | ||
A transannular approach to the synthesis of the pyrrolizidine framework was first explored by Wilson et al., who showed that electrophiles could activate 5-aza-cis-cyclooctenes toward transannular C–N bond construction.11,12 The groups of White,4 Lin,13 Madsen5 and Davies14 have elegantly demonstrated the importance of transannular cyclizations in syntheses of pyrrolizidine alkaloids. White first used an epoxidation/transannular cyclization strategy to synthesize australine, and demonstrated that the 5-aza-cis-cyclooctene precursors could be created using ring closing metathesis.4 Madsen later synthesized australine via this approach of epoxidation/transannular cyclization, and demonstrated that the 5-aza-cis-cyclooctene precursors could be prepared very easily using a zinc-mediated fragmentation of iodosugars derived from fructose or sucrose.5 Most recently, Davies described an enantioselective synthesis of 7a-epi-hyacinthacine A1 in which transannular iodoamination was a key step.14
We envisioned that access to the pyrrolizidine alkaloids could be realized through transannular hydroamination15,16 of a 5-aza-cyclooctene (Scheme 1). There are few descriptions of transannular hydroaminations of 5-aza-cis-cyclooctenes. The radical cation of 5-aza-cis-cyclooctene cyclizes in the presence of t-BuSH to pyrrolizidine in 28% yield.12 The two-stage conversion of 3-azabicyclo[3.3.l]non-6-ene to 3-aza-noradamantane proceeds in two steps via the intermediacy of an isolable HCl addition product.16 We envisioned that transannular hydroaminations of 5-aza-trans-cyclooctenes would be facile due to the intrinsic strain of the (E)-cycloalkene. While stereospecific, transannular cyclizations of (E)-cycloalkenes have been studied with larger ring systems,17 relatively few studies have been carried out on trans-cyclooctene derivatives.18–20 Cerè et al. demonstrated that (E)-thiacyclooct-4-ene undergoes acid catalyzed transannular cyclization,21 as does the anion of (E)-4,5-epoxy-1-thiacyclooctane-1,1-dioxide.22 In 2008, we demonstrated that treatment of 4-aza-trans-cyclooctene derivative with bromine provides the bromopyrrolizidine in >90% isomeric purity, and with complementary diastereoselectivity relative to bromoamination of 4-aza-cis-cyclooctene.23 Previously, the transannular hydroamination of an 5-aza-trans-cyclooctene has not been described, although elegant studies on the intermolecular hydroamination of trans-cycloalkenes have been described by Beauchemin et al.24
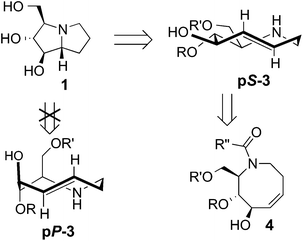 | ||
| Scheme 1 Retrosynthesis of hyacinthacine 1 using transannular hydroamination and diastereoselective photoisomerization as key steps. | ||
Retrosynthetic analysis
A retrosynthetic analysis for the total synthesis of hyacinthacine A2 (1) is displayed in Scheme 1. It was expected that 1 could arise from the hydroamination of a 5-aza-trans-cyclooctene pS-3, which would in turn arise from the photoisomerization of cis-cyclooctene 4.25cis-Cyclooctene 4 could be prepared via ring closing metathesis.4,5 We planned for the photoisomerization of 4 to be carried out on a practical scale using a flow reaction developed by our group, in which trans-isomers selectively complex to AgNO3/silica.23 While our photochemical flow procedure has been applied in bioconjugation chemistry,26,27 it has not been previously applied in a target directed synthesis.A key consideration for the synthesis of hyacinthacine A2 is stereocontrol in the photoisomerization step, as trans-cyclooctenes possess planar chirality and are configurationally stable. Only the pS isomer of 3 would lead to the natural product; pP-3 would instead lead to the 7a epimer of hyacinthacine A2. In our earlier study, poor diastereoselectivity was observed in the photoisomerizations of most cyclooctenes that bear stereogenic centers.23 For example, the photoisomerization of 4,5-dihydroxy-trans-cyclooctene 5 gives 6a and 6b with a dr of only 1.9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (Scheme 2a). An exceptional case was 7, which gave 8a and 8b in 11:1 dr (Scheme 2b). As 7 is simply the acetonide of 5, we hypothesized that the high diastereoselectivity observed in the photoisomerization of 7 was a result of conformational constraints of the ring fusion. The eight-membered rings of both diastereomers of 6 can adopt the most stable crown conformation. By contrast, only the major diastereomer of 8 is able to adopt a crown conformation. The minor diastereomer 8b cannot reasonably support a crown conformation, which would necessitate a trans-diaxial ring fusion. Compound 8b must instead adopt a higher energy conformation. The second lowest energy conformation of trans-cyclooctene is the chair conformation, which is calculated to be 5.4–5.6 kcal mol−1 (22.6–23.4 kJ mol−1) higher in energy than the crown conformation.23,28 This difference in product ground state conformations is apparently mirrored by an energetic difference in the transition states leading to 8a and 8b.
:1 (Scheme 2a). An exceptional case was 7, which gave 8a and 8b in 11:1 dr (Scheme 2b). As 7 is simply the acetonide of 5, we hypothesized that the high diastereoselectivity observed in the photoisomerization of 7 was a result of conformational constraints of the ring fusion. The eight-membered rings of both diastereomers of 6 can adopt the most stable crown conformation. By contrast, only the major diastereomer of 8 is able to adopt a crown conformation. The minor diastereomer 8b cannot reasonably support a crown conformation, which would necessitate a trans-diaxial ring fusion. Compound 8b must instead adopt a higher energy conformation. The second lowest energy conformation of trans-cyclooctene is the chair conformation, which is calculated to be 5.4–5.6 kcal mol−1 (22.6–23.4 kJ mol−1) higher in energy than the crown conformation.23,28 This difference in product ground state conformations is apparently mirrored by an energetic difference in the transition states leading to 8a and 8b.
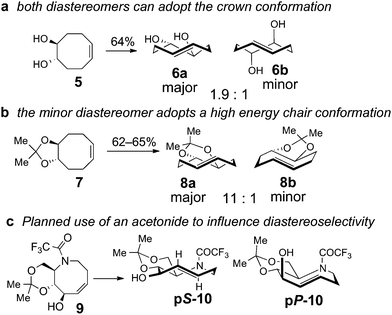 | ||
| Scheme 2 (a) Like most cyclooctenes with stereogenic centers, the photoisomerization of cyclooctene 5 proceeds with poor diastereoselectivity. (b) The high diastereoselectivity observed in photoisomerization of cyclooctene 7 is attributed to the acetonide ring fusion, which would force the minor diastereomer to adopt a high energy chair conformation. (c) Incorporation of an acetonide in 5-aza-cyclooctene 9 is expected to prefer the formation of pS-10. | ||
Based on these observations, we predicted that a ring fusion could also impart diastereocontrol in the synthesis of hyacinthacine A2. Thus, acetonide 9 was targeted, and expected to photoisomerize to pS-10, which can adopt the crown conformation (Scheme 2c). The eight-membered ring of minor diastereomer pP-10 would be unable to adopt a crown conformation, and would instead be forced to adopt a higher energy chair conformer.
Results and discussion
The synthesis of acetonide 9 was accomplished as illustrated in (Scheme 3). The preparation of ketone 11 was carried out using the efficient method previously described by Lauritsen and Madsen.5 Reductive amination of 11 was carried out using a modification of the literature procedure5 to give an inseparable 7:3 mixture of diastereomers. However, the isomers could be readily separated upon trifluoroacetylation, and the major diastereomer 12 was obtained in 30% yield. While our efforts to improve the reductive amination of 11 were unsuccessful, we considered that the brevity of the synthesis of 12 makes it an attractive intermediate. Ring closing metathesis using the 2nd generation Grubbs catalyst gave 9 in 91% yield.
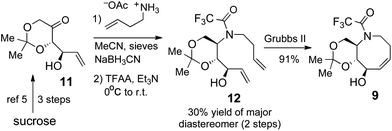 | ||
| Scheme 3 Synthesis of aza-cis-cyclooctene 9. | ||
The key photoisomerization of 9 was carried out using our previously described flow system23 in which the very unfavorable cis/trans ratio is driven to completion by active removal of the trans-isomers (Fig. 2). Thus, a 4:1 hexanes/ether solution of cis-alkene 9 was irradiated at 254 nm, and continuously cycled through a RediSep™ column that was packed with a bed of AgNO3 impregnated silica gel.29 Unlike cis-cyclooctenes, trans-cyclooctenes form stable coordination complexes with Ag(I), and trans-isomers 10 form stable complexes with AgNO3 that adhere to the column. In contrast, cis-cyclooctene 9 is not retained on the column, and is returned to the reaction flask for isomerization. The aza-trans-cyclooctene complexes were decomplexed from AgNO3 using aqueous ammonia, providing 10 with 8:1 dr, favoring the pS-isomer. The major isomer was separated, and X-ray crystallography confirmed that the eight-membered ring of pS-10 adopts a crown structure in the solid state (Fig. 2a).
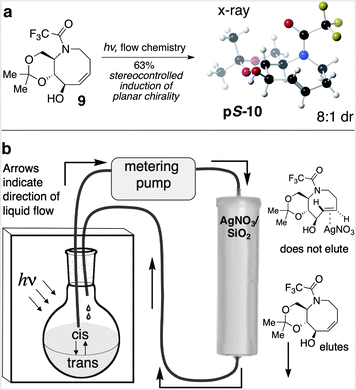 | ||
| Fig. 2 (a) Flow photoisomerization of 9 provides 10 in 8:1 dr, favoring the pS-diastereomer. (b) Schematic description of the flow photochemical apparatus. | ||
The total synthesis of hyacinthacine A2 (1) was completed as shown in Scheme 4. The trifluoroacetyl group of pS-10 was cleaved through treatment with MeLi. Without purification, treatment with hydrochloric and acetic acids removed the acetonide to give triol 13 as the ammonium salt. 5-Aza-trans-cyclooctene 13 smoothly underwent transannular hydroamination to give hyacinthacine A2 (1) upon treatment with aqueous ammonium hydroxide and pH adjustment with aqueous hydrochloric acid. In the hydroamination, the absolute planar chirality of 13 is transferred to the C-7a stereocenter of 1 with excellent fidelity: only a single diastereomer of 1 is obtained in the transannular reaction.
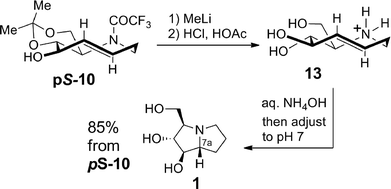 | ||
| Scheme 4 Synthesis of hyacinthacine A2 via transannular hydroamination with planar-to-point chirality transfer. | ||
Conclusions
The total synthesis of hyacinthacine A2 is reported using a novel transannular hydroamination of a 5-aza-trans-cyclooctene precursor in which planar chirality is transferred to point chirality in the product. Key to the success of this strategy was the development of a method for establishing absolute planar chirality via stereocontrolled photoisomerization of a 5-aza-cis-cyclooctene.Acknowledgements
We thank Glenn Yap for X-ray crystallography. This work was supported by NIH Grant Number GM068640-01. Spectra were obtained with instrumentation supported by NSF CRIF:MU grants: CHE 0840401 and CHE-0541775.Notes and references
- B. L. Stocker, E. M. Dangerfield, A. L. Win-Mason, G. W. Haslett and M. S. M. Timmer, Eur. J. Org. Chem., 2010, 1615–1637 CrossRef CAS; M. D. López, J. Cobo and M. Nogueras, Curr. Org. Chem., 2008, 12, 718–750 CrossRef; S. G. Pyne and M. Tang, Curr. Org. Chem., 2005, 9, 1393–1418 CrossRef; J. R. Liddell, Nat. Prod. Rep., 2002, 19, 773–781 RSC.
- Syntheses of hyacinthacine A2: L. Rambaud, P. Compain and O. R. Martin, Tetrahedron: Asymmetry, 2001, 12, 1807–1809 CrossRef CAS; F. Cardona, E. Faggi, F. Liguori, M. Cacciarini and A. Goti, Tetrahedron Lett., 2003, 44, 2315–2318 CrossRef; I. Izquierdo, M. T. Plaza and F. Franco, Tetrahedron: Asymmetry, 2003, 14, 3933–3935 CrossRef; S. Desvergnes, S. Py and Y. Vallée, J. Org. Chem., 2005, 70, 1459–1462 CrossRef; P. Dewi-Wülfing and S. Blechert, Eur. J. Org. Chem., 2006, 1852–1856 CrossRef; J. Calveras, J. Casas, T. Parella, J. Joglar and P. Clapés, Adv. Synth. Catal., 2007, 349, 1661–1666 CrossRef; C. Ribes, E. Falomir, M. Carda and J. A. Marco, Tetrahedron, 2009, 65, 6965–6971 CrossRef; I. Delso, T. Tejero, A. Goti and P. Merino, Tetrahedron, 2010, 66, 1220–1227 CrossRef; W.-J. Liu, J.-L. Ye and P.-Q. Huang, Org. Biomol. Chem., 2010, 8, 2085 Search PubMed.
- Selected syntheses of australine and alexine: S. E. Denmark and E. A. Martinborough, J. Am. Chem. Soc., 1999, 121, 3046–3056 CrossRef CAS; R. H. Furneaux, G. J. Gainsford, J. M. Mason and P. C. Tyler, Tetrahedron, 1991, 50, 2131–2160 CrossRef; W. H. Pearson and J. V. Hines, Tetrahedron Lett., 1991, 32, 5513–5516 CrossRef; W. H. Pearson and J. V. Hines, J. Org. Chem., 2000, 65, 5785–5793 CrossRef; A. Romero and C. H. Wong, J. Org. Chem., 2000, 65, 8264–8268 CrossRef; B. M. Trost, A. Aponick and B. N. Stanzl, Chem.–Eur. J., 2007, 13, 9547–9560 CrossRef; M. Dressel, P. Restorp and P. Somfai, Chem.–Eur. J., 2008, 14, 3072–3077 CrossRef; G. W. J. Fleet, M. Haraldsson, R. J. Nash and L. E. Fellows, Tetrahedron Lett., 1988, 29, 5441–5444 CrossRef; M. Takahashi, T. Maehara, T. Sengoku, N. Fujita, K. Takabe and H. Yoda, Tetrahedron, 2008, 64, 5254–5261 CrossRef; H. Yoda, H. Katoh and K. Takabe, Tetrahedron Lett., 2000, 41, 7661–7665 CrossRef.
- J. D. White and P. Hrnciar, J. Org. Chem., 2000, 65, 9129–9142 CrossRef CAS; J. D. White, P. Hrnciar and A. F. T. Yokochi, J. Am. Chem. Soc., 1998, 120, 7359–7360 CrossRef.
- A. Lauritsen and R. Madsen, Org. Biomol. Chem., 2006, 4, 2898–2905 CAS.
- N. Asano, R. J. Nash, R. J. Molyneux and G. W. J. Fleet, Tetrahedron: Asymmetry, 2000, 11, 1645–1680 CrossRef CAS; A. Kato, E. Kano, I. Adachi, R. J. Molyneux, A. A. Watson, R. J. Nash, G. W. J. Fleet, M. R. Wormald, H. Kizu, K. Ikeda and N. Asano, Tetrahedron: Asymmetry, 2003, 14, 325–331 CrossRef.
- D. L. Taylor, R. Nash, L. E. Fellows, M. S. Kang and A. S. Tyms, Antiviral Chem. Chemother, 1992, 3, 273–277 CAS.
- N. Asano, H. Kuroi, K. Ikeda, H. Kizu, Y. Kameda, A. Kato, I. Adachi, A. A. Watson, R. J. Nash and G. W. J. Fleet, Tetrahedron: Asymmetry, 2000, 11, 1–8 CrossRef CAS.
- J. E. Tropea, R. J. Molyneux, G. P. Kaushal, Y. T. Pan, M. Mitchell and A. D. Elbein, Biochemistry, 1989, 28, 2027–2034 CrossRef CAS.
- R. J. Nash, L. E. Fellows, J. V. Dring, G. W. J. Fleet, A. Girdhar, N. G. Ramsden, J. M. Peach, M. P. Hegarty and A. M. Scofield, Phytochemistry, 1990, 29, 111–114 CrossRef CAS.
- S. R. Wilson, R. A. Sawicki and J. C. Huffman, J. Org. Chem., 1981, 46, 3887–3891 CrossRef CAS; S. R. Wilson and R. A. Sawicki, J. Heterocycl. Chem., 1982, 19, 81–83 CrossRef.
- For transannular cyclizations involving nitrogen-centered radicals: M. Newcomb, D. J. Marquardt and T. M. Deeb, Tetrahedron, 1990, 46, 2329–2344 CrossRef CAS.
- T. Subramanian, C.-C. Lin and C.-C. Lin, Tetrahedron Lett., 2001, 42, 4079–4082 CrossRef CAS.
- E. A. Brock, S. G. Davies, J. A. Lee, P. M. Roberts and J. E. Thomson, Org. Lett., 2011, 13, 1594–1597 CrossRef CAS.
- Hydroamination review: T. E. Müller, K. C. Hultzsch, M. Yus, F. Foubelo and M. Tada, Chem. Rev., 2008, 108, 3795–3892 CrossRef CAS . Also see: B. M. Cochran and F. E. Michael, J. Am. Chem. Soc., 2008, 130, 2786–2792 CrossRef . For a transannular, Pd-catalyzed hydroamination to form tropenes: N. Sakai, A. Ridder and J. F. Hartwig, J. Am. Chem. Soc., 2006, 128, 8134–8135 CrossRef.
- J. H. Udding, N. Papin, H. Hiemstra and W. N. Speckamp, Tetrahedron, 1994, 50, 8853 CrossRef CAS.
- U. Nubbemeyer, Eur. J. Org. Chem., 2001, 1801–1816 CrossRef CAS; E. D. Edstrom, J. Am. Chem. Soc., 1991, 113, 6690–6692 CrossRef; A. Sudau, W. Münch, U. Nubbemeyer and J. W. Bats, J. Org. Chem., 2000, 65, 1710–1720 CrossRef; S. Surprenant and W. D. Lubell, Org. Lett., 2006, 8, 2851–2854 CrossRef.
- Recent studies on E-silacycloalkenes: M. Prévost and K. A. Woerpel, J. Am. Chem. Soc., 2009, 131, 14182–14183 CrossRef CAS; M. Prévost, J. W. Ziller and K. A. Woerpel, Dalton Trans., 2010, 39, 9275–9275 RSC; C. C. Ventocilla and K. A. Woerpel, J. Am. Chem. Soc., 2011, 133, 406–408 CrossRef.
- Recent studies on formation and conformational control in E-cyclononenes: K. Tomooka, N. Komine, D. Fujiki, T. Nakai and S.-i. Yanagitsuru, J. Am. Chem. Soc., 2005, 127, 12182–12183 CrossRef CAS; K. Tomooka, M. Suzuki, M. Shimada, S.-i. Yanagitsuru and K. Uehara, Org. Lett., 2006, 8, 963–965 CrossRef; K. Tomooka, T. Akiyama, P. Man and M. Suzuki, Tetrahedron Lett., 2008, 49, 6327–6329 CrossRef; K. Tomooka, K. Uehara, R. Nishikawa, M. Suzuki and K. Igawa, J. Am. Chem. Soc., 2010, 132, 9232–9233 CrossRef.
- Recent total synthesis of rhazinilam with transfer of axial-to-point chirality: Z. Gu and A. Zakarian, Org. Lett., 2010, 12, 4224–4227 CrossRef CAS.
- V. Cerè, F. Peri, S. Pollicino and A. Antonio, J. Chem. Soc., Perkin Trans. 2, 1998, 977–980 RSC.
- V. Cerè, C. Paolucci, S. Pollicino, E. Sandri and A. Fava, J. Org. Chem., 1991, 56, 4513–4520 CrossRef; V. Cerè, C. Paolucci, S. Pollicino, E. Sandri and A. Fava, J. Org. Chem., 1992, 57, 1457–1461 CrossRef.
- M. Royzen, G. P. A. Yap and J. M. Fox, J. Am. Chem. Soc., 2008, 130, 3760–3761 CrossRef CAS.
- J. Moran, P. H. Cebrowski and A. M. Beauchemin, J. Org. Chem., 2008, 73, 1004–1007 CrossRef CAS.
- R. Maeda, T. Wada, T. Mori, S. Kono, N. Kanomata and Y. Inoue, J. Am. Chem. Soc., 2011, 133, 10379–10381 CrossRef CAS; Y. Inoue, T. Yokoyama, N. Yamasaki and A. Tai, J. Am. Chem. Soc., 1989, 111, 6480–6482 CrossRef; N. Yamasaki, Y. Inoue, T. Yokoyama and A. Tai, J. Photochem. Photobiol., A, 1989, 48, 465–465 CrossRef; Y. Inoue, N. Yamasaki, T. Yokoyama and A. Tai, J. Org. Chem., 1993, 58, 1011–1018 CrossRef; Y. Inoue, E. Matsushima and T. Wada, J. Am. Chem. Soc., 1998, 120, 10687–10687 CrossRef; T. Inoue, K. Matsuyama and Y. Inoue, J. Am. Chem. Soc., 1999, 121, 9877–9877 CrossRef . For a recent review on the use of photochemistry in natural product synthesis, see: T. Bach and J. P. Hehn, Angew. Chem., Int. Ed., 2011, 50, 1000–1045 CrossRef.
- M. T. Taylor, M. L. Blackman, O. Dmitrenko and J. M. Fox, J. Am. Chem. Soc., 2011, 133, 9646–9649 CrossRef CAS.
- R. Selvaraj, S. Liu, M. Hassink, C.-w. Huang, L.-p. Yap, R. Park, J. M. Fox, Z. Li and P. S. Conti, Bioorg. Med. Chem. Lett., 2011 DOI:10.1016/j.bmcl.2011.04.116; Z. Li, H. Cai, M. Hassink, M. L. Blackman, R. C. D. Brown, P. S. Conti and J. M. Fox, Chem. Commun., 2010, 46, 8043–8043 RSC; M. L. Blackman, M. Royzen and J. M. Fox, J. Am. Chem. Soc., 2008, 130, 13518–13519 CrossRef CAS.
- R. D. Bach, J. Am. Chem. Soc., 2009, 131, 5233–5243 CrossRef CAS.
- C. M. Williams and L. N. Mander, Tetrahedron, 2001, 57, 425–447 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental and characterization details, 1H and 13C NMR spectra for new compounds, and the CIF file for pS-10. CCDC reference number 833725. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c1sc00442e |
| This journal is © The Royal Society of Chemistry 2011 |