A trigonal and hindered tertiary phosphine ligand rendered anionic by a niobate anchor: Formation of zwitterionic M(I) (M = Cu, Ag, Au, Rh) complexes†
Sidney E.
Creutz
,
Ivo
Krummenacher
,
Christopher R.
Clough
and
Christopher C.
Cummins
*
Department of Chemistry, Massachusetts Institute of Technology, Cambridge, MA, USA 02139-4307. E-mail: ccummins@mit.edu
First published on 16th August 2011
Abstract
Treatment of terminal phosphide anion [PNb(N[Np]Ar)3]− ([Na(OEt2)]+ salt, Ar = 3,5-Me2C6H3) with two diphenylketene equivalents led to isolation of anion [P(C{CPh2}O)2Nb(N[Np]Ar)3]− in 78% yield as its [Na(THF)]+ salt. The latter reacts with diphenylketene (1 equiv) to provide the triple diphenylketene addition product [P(C{CPh2}O)3Nb(N[Np]Ar)3]− in 88% yield as its [Na(THF)]+ salt; the same material is obtained alternatively in 93% yield by reaction of diphenylketene (3 equiv) directly with niobium phosphide [Na(OEt2)][PNb(N[Np]Ar)3]. The anion [P(C{CPh2}O)3Nb(N[Np]Ar)3]− was also crystallized as its ion-separated [Na(THF)6]+ salt as illuminated by a single-crystal X-ray diffraction study, which also revealed the three-fold symmetric nature of the highly hindered tertiary phosphine comprising the anionic component. Coinage metal monocations of the new, anionic phosphine were prepared via salt elimination; structural studies on the zwitterionic complexes (py)M[P(C{CPh2}O)3Nb(N[Np]Ar)3] (M = Cu, Ag, and Au) showed them to be isostructural in the space groupP21/c while illustrating the ensconcement of a two-coordinate coinage metal in a deep binding pocket flanked by three phenyl residues. Rhodium(I) incorporation into the anion's binding pocket illustrated versatility of the latter: in contrast to structural observations for the M(I) complexes (M = Cu, Ag, and Au), in the case of Rh[P(C{CPh2}O)3Nb(N[Np]Ar)3] an X-ray study reveals strong interactions (η6 and η2, respectively) with two of the three phenyl residues that are proximal to Rh(I) when connected to the phosphine lone pair of the anion.
Introduction
Strategies for the development of negatively charged phosphine ligands have included sulfonation for aqueous applications,1 and also the replacement of carbon with boron in an otherwise familiar organic phosphine ligand structure. The latter strategy was elegantly implemented with the synthesis of diphenylphosphidoboratabenzene (DPB) as an isosteric but negatively charged analog of PPh3.2 Substitution with boron of the axial bridgehead carbon of the well-known triphos ligand,3 has given rise to a tridentate triphosphine bearing a single negative charge.4 Zwitterionic complexes of related anionic bidentate diphosphine ligands have been studied for their catalytic activity in comparison with analogous cationic complexes.5 Tethering of the tetraphenylborate ion to a phosphine ligand structure has given rise also to a class of anionic phosphine ligands known as (phosphino)tetraphenylborates.6 Of course, interest in negatively charged phosphine ligands stems from the fact that charge-neutral molecules incorporating them may have solubility in low-dielectric organic media that is improved over their cationic counterparts, together with the potential for such ligands to exhibit enhanced electron donicity.6 Furthermore, having a counter-ion that is built in to a ligand framework assures a high level of control over its interaction with a cationic metal center,7 thus representing an alternative approach to problems normally addressed via weakly coordinating anions.8 Weakly coordinating anions themselves most frequently appear in the guise of fluorinated arylborates such as [B(3,5-C6H3(CF3)2)4]− or [B(C6F5)4]−,9,10 or as halogenated carboranes,8 but examples based on six-coordinate niobium anions, “niobates”, e.g. [Nb(OC6F5)6]− also are known.11 With the present work we show that it is possible efficiently to assemble a crowded tertiary phosphine ligand that is niobate-anchored, simply via triple addition of diphenylketene to the terminal phosphide anion [P![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) Nb(N[Np]Ar)3]− (1, Ar = 3,5-Me2C6H3 and Np = neopentyl). Raturi et al. have described a related cascade synthesis of a three-fold symmetric phosphine metalloligand.12
Nb(N[Np]Ar)3]− (1, Ar = 3,5-Me2C6H3 and Np = neopentyl). Raturi et al. have described a related cascade synthesis of a three-fold symmetric phosphine metalloligand.12
Investigations into the reactivity of the niobium terminal phosphide functional group exemplified within anion 1 have centered on P-functionalization and transfer initiated by treatment with an electrophile.13 For example, treatment of [Na(OEt2)][1] with pivaloyl chloride gave, after initial NaCl elimination, an isolable metallacycle that was subject to thermal fragmentation producing known phosphaalkyne tBuCP14 together with the terminal oxo ONb(N[Np]Ar)3.13 In none of these studies did the addition of multiple equivalents of a given electrophile to 1 become manifest. With the present contribution we show that diphenylketene is a special electrophile in this regard, possessing the capability to undergo double or even triple addition to the terminal phosphide anion complex 1.
A key finding of the present work is that triple diphenylketene addition to phosphide 1 assembles an unusual negatively charged and three-fold symmetric metal ion binding pocket. An impressive structural attribute of the new metalloligand15 is the rigid positioning—in a picket fence16-like array—of three phenyl residues about the metal binding site. These phenyl residues may simply surround and prevent lateral access to a metal lodged in the binding site as demonstrated below for the d10 metal ions copper(I), silver(I), and gold(I), or, alternatively, they may become strongly interacting as we find to be the case for rhodium(I).
Results and discussion
While addition of one equivalent of diphenylketene to [Na(OEt2)][1] led to an ill-defined system with broad NMR features, the use of two diphenylketene equivalents gave a system amenable both to complete spectroscopic characterization and isolation as dark red crystals. Accordingly, the benzene-soluble salt [Na(THF)][P(C{CPh2}O)2Nb(N[Np]Ar)3] ([Na(THF)][2]) was isolated in 78% yield and has a characteristic 31P NMR signal located at δ = −85 ppm.The molecular structure of [Na(THF)][2] (see Fig. 1) is dominated by a tight ion pair involving electrostatic association of the sodium cation with sources of electron density in the anion, including close contacts with an aryl residue of one of the anilide ligands, and interactions with enolate carbon and oxygen atoms (O1, O2, C1, and C3).19 Also providing sodium ion solvation is the single THF molecule that is a rigorous component of the molecular formula, consistent with combustion analysis data. Notably absent is any interaction between the phosphorus atom P1 and the sodium cation. In addition, the niobium-phosphorus interatomic distance of 2.6419(7) Å is somewhat long as compared with the sum of single-bond covalent radii for these elements: r(Nb, 1.47 Å) + r(P, 1.11 Å) = 2.58 Å.20 A search of the Cambridge Structural Database for niobium-phosphorus distances in the 2.6–2.7 Å range returned 110 hits, with the vast majority of these being tertiary phosphine complexes; two of the hits were however phosphanide (Nb–PR2) systems with pyramidal P atoms and what must be considered bona fideniobium-phosphorus single bonds; an example is the complex Cp2Nb(PPh2)(HPPh2).21Niobium complexes of P-unsaturated ligands are also found in this distance range. For example, a beautiful complex containing the cyclo-P4 ligand fits this description: Cp*Nb(η4-P4)(CO)2.22 Also of this ilk is a series of dinuclear complexes with bridging cyclo-EP2 (E = Ge, Sn, Pb) ligands,23 together with complexes of the diphosphene PhP![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) PPh as well as of the parent, HPPH.24 For reference, the NbP triple bond in anion 1 is associated with a short interatomic distance: 2.186(2) Å.13
PPh as well as of the parent, HPPH.24 For reference, the NbP triple bond in anion 1 is associated with a short interatomic distance: 2.186(2) Å.13
![platon
17-generated ORTEP18 representation of [Na(THF)][2] with ellipsoids at the 50% probability level. Selected interatomic distances (Å) and angles (°): Nb1 N2 2.005(2), Nb1 N1 2.040(2), Nb1 N3 2.046(2), Nb1 O1 2.1325(18), Nb1 O2 2.1683(18), Nb1 P1 2.6419(7), Na1 O1S 2.216(2), Na1 O2 2.270(2), Na1 O1 2.492(2), Na1 C16 2.617(3), Na1 C3 2.656(3), Na1 C11 2.820(3), Na1 C15 2.911(3), Na1 C1 2.950(3), P1 C1 1.862(3), P1 C3 1.869(2), C1 O1 1.359(3), C1 C2 1.363(4), O2 C3 1.350(3), C4 C3 1.362(4).](/image/article/2011/SC/c1sc00375e/c1sc00375e-f1.gif) | ||
| Fig. 1 PLATON 17-generated ORTEP18 representation of [Na(THF)][2] with ellipsoids at the 50% probability level. Selected interatomic distances (Å) and angles (°): Nb1 N2 2.005(2), Nb1 N1 2.040(2), Nb1 N3 2.046(2), Nb1 O1 2.1325(18), Nb1 O2 2.1683(18), Nb1 P1 2.6419(7), Na1 O1S 2.216(2), Na1 O2 2.270(2), Na1 O1 2.492(2), Na1 C16 2.617(3), Na1 C3 2.656(3), Na1 C11 2.820(3), Na1 C15 2.911(3), Na1 C1 2.950(3), P1 C1 1.862(3), P1 C3 1.869(2), C1 O1 1.359(3), C1 C2 1.363(4), O2 C3 1.350(3), C4 C3 1.362(4). | ||
In Scheme 1, compound [Na(THF)][2] is drawn with a P–Nb single bond and a resulting formal negative charge at niobium. Another limiting form would be to draw a dative P→Nb interaction with the negative charge placed on phosphorus. Such an alternative representation is certainly evocative of the subsequent nucleophilic reactivity of [Na(THF)][2] at the phosphorus center.
![Synthesis of complex salts [M][2], and [M][3].](/image/article/2011/SC/c1sc00375e/c1sc00375e-s1.gif) | ||
| Scheme 1 Synthesis of complex salts [M][2], and [M][3]. | ||
As shown in Scheme 1, addition of 1 equiv of diphenylketene to [Na(THF)][2] leads readily to the triple addition product, [M][3], which has been crystallized for an X-ray structure determination with an [Na(THF)6]+ counter-ion. On the other hand, standard work-up gives rise to loss of THF such that the counter-ion reduces to [Na(THF)]+ upon standard work-up and drying under vacuum, as determined by NMR integration. Given that [Na(THF)]+ is tightly associated in the solid-state structure of [Na(THF)][2], such association is also very likely in the case of [Na(THF)][3], and possibly the sodium ion in this case occupies the trigonal binding site where it would interact with the phosphorus lone pair.
For [Na(THF)][3], the 31P NMR shift is −23 ppm in benzene-d6 and the signal is broad (Δν1/2 = 105 Hz). In THF-d8 the signal is sharp and appears at −34 ppm. In pyridine the 31P NMR shift is −30 ppm, while in dichloromethane a sharp resonance is found at −34 ppm. The solvent-dependence of the NMR data for [Na(THF)][3] suggests that in the case of benzene as solvent, the sodium ion resides in the trigonal binding pocket where it may interact with phosphorus (vide infra), whereas in more polar or coordinating solvents (pyridine, THF, dichloromethane) the sodium ion is extracted from the binding pocket.
That the binding pocket of anion [3]− may be referred to as trigonal is underscored by the fact that its [Na(THF)6]+ salt crystallizes in the space groupR![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) with both niobium and phosphorus atoms located on the crystallographic three-fold axis of symmetry (see Fig. 2).
with both niobium and phosphorus atoms located on the crystallographic three-fold axis of symmetry (see Fig. 2).
![platon
17-generated ORTEP18 representation of anion [3]− (the non-interacting [Na(THF)6]+ counter-ion is not shown) with ellipsoids at the 50% probability level. Selected interatomic distances (Å) and angles (°): Nb1 O1 2.0507(15), Nb1 N1 2.0596(18), P1 C1 1.864(2), O1 C1 1.331(3), C1 C2 1.368(3), O1 Nb1 O1 82.16(6), O1 Nb1 N1 92.35(7), O1 Nb1 N1 87.06(7), O1 Nb1 N1 168.47(7), N1 Nb1 N1 97.51(6) C1 P1 C1 96.49(10), C1 O1 Nb1 132.73(14), O1 C1 P1 118.35(16).](/image/article/2011/SC/c1sc00375e/c1sc00375e-f2.gif) | ||
| Fig. 2 PLATON 17-generated ORTEP18 representation of anion [3]− (the non-interacting [Na(THF)6]+ counter-ion is not shown) with ellipsoids at the 50% probability level. Selected interatomic distances (Å) and angles (°): Nb1 O1 2.0507(15), Nb1 N1 2.0596(18), P1 C1 1.864(2), O1 C1 1.331(3), C1 C2 1.368(3), O1 Nb1 O1 82.16(6), O1 Nb1 N1 92.35(7), O1 Nb1 N1 87.06(7), O1 Nb1 N1 168.47(7), N1 Nb1 N1 97.51(6) C1 P1 C1 96.49(10), C1 O1 Nb1 132.73(14), O1 C1 P1 118.35(16). | ||
It is well known that both the basicity25 and nucleophilicity26 of a tertiary phosphine are affected by the C–P–C bond angles, owing to the fact that formation of a fourth bond to phosphorus requires less rehybridization the closer these angles are to the tetrahedral angle in the starting phosphine. In terms of the structural environment at phosphorus, anion [3]− contains phosphorus at the bridgehead position of a bicyclic-[2.2.2] core in which all P–C bonds involve sp2-hybridized carbon atoms. This is most similar to the case of phosphatriptycene (PT)27 and diphosphatriptycene (DPT),28 for which the C–P–C angles are respectively 91° and 97°. For comparison, anion [3]− exhibits a C1–P1–C1′ angle of 96.49(10)°, such that the similarity of its phosphorus environment to that of DPT is likely quite strong. The appropriate reference compound, triphenylphosphine, has a significantly larger C–P–C angle of 103°.29 The expectation in the case of caged phosphine structures such as that in PT, DPT, and in anion [3]−, is that since the ability of P to form tetrahedral C–P–C angles is restricted by the cage, a fourth bond formed to P will be exceptionally rich in s character.
Jongsma and Bickelhaupt showed that the reactivity of PT is diminished relative to that of triphenylphosphine, and they also used the equations of Parks30 to calculate a C–P–C bond angle of PT (93°) from its 31P NMR chemical shift of −64.8 ppm.27 It is typical for cage-constrained phosphines with constricted C–P–C angles to have shifts upfield of that for PPh3 (−3.1 ppm in C6D6). For example, the 9-phospha-10-silatriptycene P(C6H4)3SiMe (PSiT) has its 31P NMR signal at −44.4 ppm.31 The related 8-phospha-4-silathiophenetriptycene displays its 31P NMR signal at −92.15 ppm.32
The combination of [Na(THF)][3] with group 11, d10 precursors led to the formation of two-coordinate complexes containing gold(I), silver(I), or copper(I) coordinated to phosphorus within the trigonal binding pocket (Scheme 2). In each case, the metal's essentially linear coordination environment is completed by a second axial donor ligand derived either from the metal precursor or from solvent. Because these reactions proceed via salt elimination, the group 11 complexes of anion 3 so obtained are neutral zwitterionic entities. For example, treatment of [Na(THF)][3] with Au(THT)X (THT = tetrahydrothiophene; X = Br, Cl) in thawing THF resulted in rapid formation of (THT)Au[3] and precipitation of NaX. It should be noted that Au(THT)X is commonly employed as a precursor to gold(I) complexes in order to take advantage of the THT ligand lability,33–35 in distinct contrast with the salt-elimination pathway favored in the reaction with [Na(THF)][3].
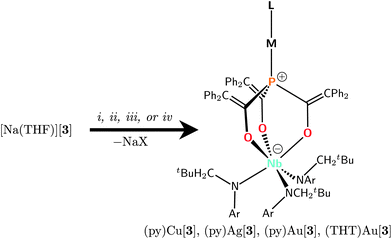 | ||
| Scheme 2 Conditions for installing group 11 d10 metal ions into the binding pocket: i, CuCl, toluene/pyridine, 60°, 1 h, 80%; ii, AgOTf, pentane/pyridine, frozen →25°, 2 min, 86%; iii, Au(THT)Br, THF/pyridine, frozen →25°, 3 h, 85%; iv, Au(THT)Br, THF, frozen →25°, 3 h, 55%. | ||
On the other hand, the THT ligand of (THT)Au[3] did prove to be substitutionally labile, such that it could be lost in favor of pyridine by dissolving (THT)Au[3] in pyridine to give (py)Au[3]. This lability probably contributes to the relatively low isolated yield of (THT)Au[3] (55% for X = Br); with this in mind, it was expected that the direct formation of (py)Au[3] by treatment of [Na(THF)][3] with Au(THT)Br in thawing pyridine as the solvent, without the intermediate isolation of (THT)Au[3], would give a higher yield. Indeed, this protocol led to an 85% isolated yield of the desired pyridine complex.
Both (L)Au[3] (L = THT, py) complexes have 31P NMR shifts downfield of the free phosphine (δ = 16 and 7 ppm, respectively), as expected for metal complexation to a phosphine.36 The 1H NMR (benzene-d6, 20 °C, 500 MHz) spectra of these complexes reveal that they maintain C3 symmetry, indicative of rapid rotation about the Au–L bond, though the THT resonances are broad. However, the phenyl rings surrounding the binding pocket exhibit restricted rotation; the ortho and metaprotons on each ring are chemically inequivalent on the NMR timescale. An upfield shift for the phenyl1H and 13C nuclei would be expected in case of significant Au⋯π interaction,36 but this is not observed.
The gold complexes have been structurally characterized by single-crystal X-ray diffraction following crystal growth by vapor diffusion of pentane into THF (Fig. 3). The metrical parameters of the phosphine metalloligand are not significantly different from those obtained for [Na(THF)6][3]. The Au–S interatomic distance is lengthened by about 0.08 Å relative to solid-state Au(THT)Cl (which occurs as a polymeric chain with Au–Au bonds).37 The closest distance between the gold atoms and any of the phenylipsocarbon atoms surrounding the binding pocket is 3.138 Å for L = THT and 3.072 Å for L = py, these being much longer than the bond lengths which have been computationally determined38,39 or experimentally measured40–42 for strong gold-arene interactions. Additionally, it has been suggested that the longest bond distance for a significant gold-arene interaction is 2.95 Å.40 Nevertheless, our observed Au–C distances may be consistent with a weak gold-arene interaction, since the distances observed are less than the sum of the Van der Waals radii of the atoms, and several complexes reported to have such interactions evinced similar distances.40,43–45 However, it is also possible that the relatively close approach of the gold cation to the phenyl rings is primarily enforced by ligand structural constraints rather than electronic considerations.
![platon
17-generated ORTEP18 representation of the gold pyridine complex (py)Au[3] with ellipsoids at the 50% probability level. The copper and silver derivatives (py)Cu[3] and (py)Ag[3] similarly crystallized with 2.5 molecules of THF (not shown) in the space groupP21/c. Selected interatomic distances (Å) and angles (°) for (py)Au[3]: Au1 N4 2.091(4), Au1 P1 2.2443(12), Nb1 N1 2.028(4), Nb1 N3 2.030(4), Nb1 N5 2.032(4), Nb1 O3 2.094(3), Nb1 O1 2.095(3), Nb1 O2 2.108(3), C70 C71 1.365(6), C81 C80 1.369(6), C91 C90 1.360(6), N4 Au1 P1 173.89(13), C80 P1 C90 100.0(2), C80 P1 C70 98.1(2), C90 P1 C70 102.5(2); for (py)Cu[3]: Cu1 N4 1.931(2), Cu1 P1 2.1757(6), N4 Cu1 P1 172.43(6); for (py)Ag[3]: Ag1 N4 2.162(3), Ag1 P1 2.3678(9), N4 Ag1 P1 169.13(8).](/image/article/2011/SC/c1sc00375e/c1sc00375e-f3.gif) | ||
| Fig. 3 PLATON 17-generated ORTEP18 representation of the gold pyridine complex (py)Au[3] with ellipsoids at the 50% probability level. The copper and silver derivatives (py)Cu[3] and (py)Ag[3] similarly crystallized with 2.5 molecules of THF (not shown) in the space groupP21/c. Selected interatomic distances (Å) and angles (°) for (py)Au[3]: Au1 N4 2.091(4), Au1 P1 2.2443(12), Nb1 N1 2.028(4), Nb1 N3 2.030(4), Nb1 N5 2.032(4), Nb1 O3 2.094(3), Nb1 O1 2.095(3), Nb1 O2 2.108(3), C70 C71 1.365(6), C81 C80 1.369(6), C91 C90 1.360(6), N4 Au1 P1 173.89(13), C80 P1 C90 100.0(2), C80 P1 C70 98.1(2), C90 P1 C70 102.5(2); for (py)Cu[3]: Cu1 N4 1.931(2), Cu1 P1 2.1757(6), N4 Cu1 P1 172.43(6); for (py)Ag[3]: Ag1 N4 2.162(3), Ag1 P1 2.3678(9), N4 Ag1 P1 169.13(8). | ||
Complexes of the phosphine metalloligand [3] with silver and copper show generally similar characteristics to the gold complexes, and are essentially isostructural in the solid state. (py)Ag[3] was formed in 86% yield via the treatment of [Na(THF)][3] with AgOTf in pyridine solvent. As found for the gold complexes, the proton NMR spectrum of (py)Ag[3] is indicative of C3 symmetry and hindered rotation of the phenyl groups. Also, the pyridine resonances are somewhat broadened, perhaps indicative of hindered rotation about the Ag-N bond. The 31P-NMR spectrum of (py)Ag[3] contains a sharp resonance at −8 ppm; this peak is split into two doublets with well resolved coupling to both 107Ag and 109Ag (1JAg–P = 670 Hz, 780 Hz). These coupling constants are relatively large compared to those reported for simple silver(I) phosphine adducts, but they are still well within reported ranges. Interestingly, there is also silver-carbon coupling evident in the 13C NMR involving the ipso carbons of the phenyl rings surrounding the binding pocket (1JAg–C = 1.1 Hz). Since there is no coupling to the benzylic carbon, the observed JAg–C coupling must result from through-space interactions directly between silver and the ipso carbons, rather than through the covalent bonding network.
Stirring [Na(THF)][3] with CuCl in the presence of one equivalent of pyridine (60°, 1 h) resulted in the formation of (py)Cu[3], which was isolated in 80% yield as a bright yellow solid. No conversion was observed either in the absence of pyridine or in the presence of a large excess of pyridine, most likely because the copper cations were sequestered as [Cu(py)n]+ species.47 The 31P NMR shift of (py)Cu[3] is −10 ppm. The 1H NMR spectrum of (py)Cu[3] is consistent with C3 symmetry, and the resonances for the “up” phenyl rings are only slightly broadened due to hindered rotation. This suggests that phenyl rotation is somewhat more facile in this species than in the analogous silver and gold complexes, consistent with diminished steric congestion in (py)Cu[3] stemming from the smaller size of the copper(I) ion. There is a single, sharp set of three pyridine resonances, indicating rapid rotation of the pyridine moiety within the binding pocket, also consistent with less steric congestion than in the gold and silver congeners.
The closest approaches of the silver and copper cations to the phenyl ring carbon atoms in (py)M[3] are respectively 3.032 Å and 2.921 Å; the latter values are not sufficiently short to be indicative of strong metal-arene interactions.40,48 In the case of silver, the Ag⋯Cipso distance is barely within the range which has been reported for weak silver(I)-arene interactions,40 while the Cu⋯Cipso distance is substantially longer than reported bond lengths for copper(I)-arene interactions. The observed trend in the (py)M[3] metal-arene distances is as expected solely on the basis of the ionic radii (Au > Ag > Cu).
Thus, in its complexes with gold, silver, and copper, anion [3] acts essentially as an extremely bulky phosphine. The extent to which the ligand envelops and protects the metal ions can be seen most clearly in space-filling models (Fig. 4). Estimated from the crystal structures of these compounds, the Tolman cone angle49 ranges from 270° for (py)Cu[3] to 286° for (py)Au[3], such that the ligand occupies approximately three quarters of the coordination sphere for these metals.
![Spacefilling CPK model of (py)Au[3] generated using Jmol.46](/image/article/2011/SC/c1sc00375e/c1sc00375e-f4.gif) | ||
| Fig. 4 Spacefilling CPK model of (py)Au[3] generated using Jmol.46 | ||
Treatment of [Na(THF)][3] with [Rh(COD)Cl]2 (COD = 1,5-cyclooctadiene) in the presence of 1 equiv pyridine (toluene, 60 °C) led to the formation of Rh[3] in 74% yield (Scheme 3). It was determined that the presence of pyridine (1 equiv) is necessary for the formation of this complex; in the absence of pyridine or in the presence of excess pyridine the reaction does not proceed even upon heating to 90 °C. Pyridine's role may include breaking up the dimeric starting material [Rh(COD)Cl]2 to generate monomeric [RhCl(COD)(py)], which is analogous to known complexes [RhCl(COD)(py′)], where py′ represents a substituted pyridine.50 Furthermore, it may be that the complex (py)Rh[3] is formed en route to the final, isolated product, Rh[3]. Monitoring of this reaction system by 1H NMR indeed revealed the formation of an intermediate prior to the ultimate formation of Rh[3]. In the absence of heating, the starting material is completely converted to this intermediate within ten minutes of the addition of [Rh(COD)Cl]2 and pyridine. The 1H NMR and 31P NMR spectra for this intermediate are very similar to those for [Na(THF)][3], with the phosphorus chemical shift appearing approximately 10 ppm upfield of that for the starting material; no rhodium-phosphorus coupling is evident.
![Synthesis of complex Rh[3].](/image/article/2011/SC/c1sc00375e/c1sc00375e-s3.gif) | ||
| Scheme 3 Synthesis of complex Rh[3]. | ||
The crystal structure of Rh[3] clearly shows strong interactions between the rhodium cation and the π systems of two of the phenyl rings surrounding the binding pocket of [3] (Fig. 5). Rhodium coordinates to one phenyl ring in an η6 fashion and to a second phenyl ring in an η2 fashion; there is no interaction with the third phenyl ring. The C–C distances within the rings corroborate these interactions; in the uncoordinated phenyl ring, the observed range is from 1.383(4) to 1.397(3) Å. In the η6-coordinated phenyl ring, the Cipso–Cortho distances are substantially lengthened to 1.432(3) and 1.439(3) Å, while the other bonds in the ring are only very slightly lengthened. As is suggested by these bond lengths, the coordination to the η6 ring is slightly off-center, with rhodium more closely approaching the ipsocarbon (Rh–C 2.195(2) Å) than the paracarbon (Rh–C 2.415(2) Å). Moreover, the coordination is also slightly laterally asymmetric, with Rh-Cmeta bond distances of 2.428(2) and 2.400(2) Å. The average Rh–C bond length, 2.34 Å, is in a range typical of aromatic rings η6-coordinated to rhodium(I).51–54 The C–C distances in the η2-coordinated phenyl ring show striking evidence of the disruption of the ring's aromaticity due to rhodium binding. The coordinated alkene moiety (C816–C811) has a lengthened bond, and the C812–C813 and C814–C815 interatomic distances are indicative of double bond character arising from loss of aromaticity.55,56 Additionally, the bonding environment of the phosphorus atom is considerably distorted, with the rhodium cation substantially displaced from the phosphinepseudo-C3 axis; the Nb–P–Rh angle is approximately 159°. Furthermore, at 2.1643(6) Å the Rh–P interatomic distance is short as compared with what is typically seen for rhodium(I) phosphine systems.52,53,57,58
![platon
17-generated ORTEP18 representation of Rh[3] with ellipsoids at the 50% probability level. Selected interatomic distances (Å): Rh1 P1 2.1643(6), Rh1 C711 2.195(2), Rh1 C811 2.214(2), Rh1 C816 2.237(2), Rh1 C716 2.290(2), Rh1 C712 2.322(2), Rh1 C713 2.400(2), Rh1 C714 2.415(2), Rh1 C715 2.428(2), C811 C812 1.448(3), C812 C813 1.365(3), C813 C814 1.424(4), C814 C815 1.359(4), C815 C816 1.431(3), C816 C811 1.443(3).](/image/article/2011/SC/c1sc00375e/c1sc00375e-f5.gif) | ||
| Fig. 5 PLATON 17-generated ORTEP18 representation of Rh[3] with ellipsoids at the 50% probability level. Selected interatomic distances (Å): Rh1 P1 2.1643(6), Rh1 C711 2.195(2), Rh1 C811 2.214(2), Rh1 C816 2.237(2), Rh1 C716 2.290(2), Rh1 C712 2.322(2), Rh1 C713 2.400(2), Rh1 C714 2.415(2), Rh1 C715 2.428(2), C811 C812 1.448(3), C812 C813 1.365(3), C813 C814 1.424(4), C814 C815 1.359(4), C815 C816 1.431(3), C816 C811 1.443(3). | ||
The 31P NMR properties of Rh[3] are unremarkable for tertiary phosphines bound to rhodium(I), with a chemical shift of 56 ppm and a rhodium-phosphorus one-bond coupling constant of 168 Hz. The 1H NMR spectrum of this complex reveals some more interesting aspects of its properties with respect to the interaction between rhodium and the phenyl rings that encircle the binding pocket. Despite this complex's lack of symmetry in the solid state, the solution 1H NMR spectrum is indicative of C3 symmetry, with all three phenyl rings chemically equivalent on the NMR timescale at 25 °C and down to at least −55 °C; broadening of the phenyl resonances consonant with loss of C3 symmetry is observed at this temperature. The 1H NMR signals for the aromatic hydrogens on the phenyl rings surrounding rhodium are, as is typical for aromatic rings coordinated to metals, shifted upfield; however, the magnitude of the shift is relatively small (ranging from 0.1 ppm for the metaprotons to 0.4 ppm for the orthoprotons) compared to the 1–2 ppm shifts commonly observed for η6-arene rhodium complexes.51,59,60 The relatively small shift is of course consistent with the fact that the ring is, on average, η6-bound to rhodium only one-third of the time.
Fluxionality in the rhodium-arene interactions of Rh[3] can be compared to that in other systems which display similar fluxionality via haptotropic migration.61 For instance, a rhodium(I) complex of corannulene, [(COE)2Rh(η6-C20H10)][PF6],62 possesses Rh–C interatomic distances similar to those found for Rh[3] and exhibits fluxionality on the NMR time scale in solution at 25 °C, with the rhodium cation migrating among the 6-membered aromatic rings; the latter fluxional process is frozen out at 0 °C. Despite the fluxional behavior of the system, metal-ring binding is quite strong such that the corannulene moiety is resilient against ligand substitution. Similar results have been observed for systems containing Cr(CO)3 complexed to polyaromatic systems, such as starphenylene.61,63 The explanation for the low-temperature fluxionality in these systems is that migration can occur without passing through a high-energy transition state where substantial dissociation of the metal cation from the arene system has occurred; similarly, in Rh[3], migration of rhodium among the three “up” phenyl rings evidently can occur without rhodium ever becoming substantially unsaturated.
Concluding remarks
Herein has been described the first investigation of ketene reactivity with respect to the terminal phosphide (MP) functional group. Diphenylketene was selected for this investigation due to its stability and ability to be isolated and stored as a pure substance as described originally by Staudinger,64 as well as the availability for it of a simple and reproducible preparative procedure.65 There is scant history of diphenylketene having the capability to insert multiple times at a single metal center. In the case of Cp2ZrMe2, such a modality leads to the bis-enolate complex Cp2Zr(OC[:CPh2]Me)2.66 As in the case of single diphenylketene addition to a nickel carbene complex,67 enolate-generating reactions of diphenylketene with metal–ligand multiple bonds have some precedent. Along these lines, single diphenylketene addition to an anionic alkylidyne complex of tungsten has been shown to proceed with enolate formation.68 In an unusual mode of ketene incorporation, addition to a titanium Nacnac system has provided a tripodal bis-imine enolate ligand framework.69 On the other hand, multiple ketene insertions into a metal–ligand multiple bond appear not to have been described previously. With the present work we show that such reactivity could be harnessed in order to generate a unique anionic, three-fold symmetric phosphine that is also a hindered and flexible metalloligand producing neutral and zwitterionic complexes of copper, silver, gold, and rhodium.
Acknowledgements
This material is based upon work supported by the National Science Foundation under CHE - 719157. For support including a gift of white phosphorus we thank Thermphos International. The authors are grateful also for support of I.K. by a Swiss National Science Foundation (SNF) fellowship. S.E.C. thanks Daniel Tofan, Dr Brandi M. Cossairt, and Dr Nicholas A. Piro for assistance with X-ray crystallography and MIT's UROP program for financial support.References
- B. Cornils and E. G. Kuntz, J. Organomet. Chem., 1995, 502, 177–186 CrossRef CAS.
- D. A. Hoic, W. M. Davis and G. C. Fu, J. Am. Chem. Soc., 1996, 118, 8176–8177 CrossRef CAS.
- (a) J. Chatt, F. A. Hart and H. R. Watson, J. Chem. Soc., 1962, 2537–2545 RSC; (b) P. Dapporto, S. Midollini, A. Orlandini and L. Sacconi, Inorg. Chem., 1976, 15, 2768–2774 CrossRef CAS.
- (a) A. A. Barney, A. F. Heyduk and D. G. Nocera, Chem. Commun., 1999, 2379–2380 RSC; (b) J. C. Peters, J. D. Feldman and T. D. Tilley, J. Am. Chem. Soc., 1999, 121, 9871–9872 CrossRef CAS; (c) I. R. Shapiro, D. M. Jenkins, J. C. Thomas, M. W. Day and J. C. Peters, Chem. Commun., 2001, 2152–2153 RSC; (d) D. M. Jenkins, T. A. Betley and J. C. Peters, J. Am. Chem. Soc., 2002, 124, 11238–11239 CrossRef CAS.
- C. C. Lu and J. C. Peters, J. Am. Chem. Soc., 2002, 124, 5272–5273 CrossRef CAS.
- (a) M. T. Beach, J. M. Walker, T. G. Larocque, J. L. Deagle, R. Wang and G. J. Spivak, J. Organomet. Chem., 2008, 693, 2921–2928 CrossRef CAS; (b) C. M. Thomas and J. C. Peters, Inorg. Chem., 2004, 43, 8–10 CrossRef CAS.
- W. E. Piers, Chem.–Eur. J., 1998, 4, 13–18 CrossRef CAS.
- C. A. Reed, Acc. Chem. Res., 1998, 31, 133–139 CrossRef CAS.
- M. Brookhart, B. Grant and A. F. Volpe, Organometallics, 1992, 11, 3920–3922 CrossRef CAS.
- W. E. Piers and T. Chivers, Chem. Soc. Rev., 1997, 26, 345–354 RSC.
- (a) Y. Sun, M. V. Metz, C. L. Stern and T. J. Marks, Organometallics, 2000, 19, 1625–1627 CrossRef CAS; (b) M. V. Metz, Y. Sun, C. L. Stern and T. J. Marks, Organometallics, 2002, 21, 3691–3702 CrossRef CAS.
- (a) R. Raturi, J. Lefebvre, D. B. Leznoff, B. R. McGarvey and S. A. Johnson, Chem.–Eur. J., 2008, 14, 721–730 CrossRef CAS; (b) I. Kuzu, I. Krummenacher, J. Meyer, F. Armbruster and F. Breher, Dalton Trans., 2008, 5836–5865 RSC.
- (a) J. S. Figueroa and C. C. Cummins, J. Am. Chem. Soc., 2004, 126, 13916–13917 CrossRef CAS; (b) J. S. Figueroa and C. C. Cummins, Angew. Chem., Int. Ed., 2004, 43, 984–988 CrossRef CAS; (c) J. S. Figueroa and C. C. Cummins, Dalton Trans., 2006, 2161–2168 RSC; (d) N. A. Piro, J. S. Figueroa, J. T. McKellar and C. C. Cummins, Science, 2006, 313, 1276–1279 CrossRef CAS; (e) N. A. Piro and C. C. Cummins, Inorg. Chem., 2007, 46, 7387–7393 CrossRef CAS; (f) N. A. Piro and C. C. Cummins, J. Am. Chem. Soc., 2008, 130, 9524–9535 CrossRef CAS; (g) N. Piro and C. Cummins, Angew. Chem., Int. Ed., 2009, 48, 934–938 CrossRef CAS.
- G. Becker, G. Gresser and W. Uhl, Z. Nat., 1981, 36B, 16–19 CAS.
- (a) G. S. Ferguson, P. T. Wolczanski, L. Parkanyi and M. C. Zonnevylle, Organometallics, 1988, 7, 1967–1979 CrossRef CAS; (b) T. A. Wark and D. W. Stephan, Organometallics, 1989, 8, 2836–2843 CrossRef CAS; (c) L. M. Slaughter and P. T. Wolczanski, Chem. Commun., 1997, 2109–2110 RSC.
- J. P. Collman, R. R. Gagne, C. Reed, T. R. Halbert, G. Lang and W. T. Robinson, J. Am. Chem. Soc., 1975, 97, 1427–1439 CrossRef CAS.
- A. L. Spek, J. Appl. Crystallogr., 2003, 7–13 CrossRef CAS.
- M. N. Burnett and C. K. Johnson, ORTEP-III: Oak Ridge Thermal Ellipsoid Plot Program for Crystal Structure Illustrations, Oak Ridge National Laboratory Report ORNL-6895, 1996 Search PubMed.
- Structure files in CIF format have been deposited with the CCDC with codes as follows: [Na(THF)][2], 789474; [Na(THF)6][3], 789473; (py)Cu[3], 789238; (py)Ag[3], 789236; (py)Au[3], 789235; (THT)Au[3], 789234; Rh[3], 789237†.
- (a) P. Pyykkö and M. Atsumi, Chem.–Eur. J., 2009, 15, 12770–12779 CrossRef; (b) P. Pyykkö and M. Atsumi, Chem.–Eur. J., 2009, 15, 186–197 CrossRef; (c) P. Pyykkö, S. Riedel and M. Patzschke, Chem.–Eur. J., 2005, 11, 3511–3520 CrossRef.
- G. I. Nikonov, D. A. Lemenovskii, K. Y. Dorogov and A. V. Churakov, Polyhedron, 1999, 18, 1159–1162 CrossRef CAS.
- O. J. Scherer, J. Vondung and G. Wolmershäuser, Angew. Chem., Int. Ed. Engl., 1989, 28, 1355–1357 CrossRef.
- J. S. Figueroa and C. C. Cummins, Angew. Chem., Int. Ed., 2005, 44, 4592–4596 CrossRef CAS.
- N. Etkin, M. T. Benson, S. Courtenay, M. J. McGlinchey, A. D. Bain and D. W. Stephan, Organometallics, 1997, 16, 3504–3510 CrossRef CAS.
- W. A. Henderson and C. A. Streuli, J. Am. Chem. Soc., 1960, 82, 5791–5794 CrossRef CAS.
- W. A. Henderson and S. A. Buckler, J. Am. Chem. Soc., 1960, 82, 5794–5800 CrossRef CAS.
- (a) C. Jongsma, J. P. de Kleijn and F. Bickelhaupt, Tetrahedron, 1974, 30, 3465–3469 CrossRef CAS; (b) F. J. M. Freijee and C. H. Stam, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1980, 36, 1247–1249 CrossRef.
- D. Schomburg and W. S. Sheldrick, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1975, 31, 2427–2431 CrossRef.
- J. J. Daly, J. Chem. Soc., 1964, 3799–3810 RSC.
- J. R. Parks, J. Am. Chem. Soc., 1957, 79, 757–757 CrossRef CAS.
- H. Tsuji, T. Inoue, Y. Kaneta, S. Sase, A. Kawachi and K. Tamao, Organometallics, 2006, 25, 6142–6148 CrossRef CAS.
- A. Ishii, T. Tsuchiya, J. Nakayama and M. Hoshino, Tetrahedron Lett., 1993, 34, 2347–2350 CrossRef CAS.
- A. S. K. Hashmi, Chem. Rev., 2007, 107, 3180–3211 CrossRef CAS.
- J. Ruiz, M. E. G. Mosquera, G. García, E. Patrón, V. Riera, S. García-Granda and F. V. der Maelen, Angew. Chem., Int. Ed., 2003, 42, 4767–4771 CrossRef CAS.
- R. Uson, A. Laguna, M. Laguna, B. R. Manzano, P. G. Jones and G. M. Sheldrick, J. Chem. Soc., Dalton Trans., 1985, 2417–2420 RSC.
- R. H. Crabtree, The organometallic chemistry of the transition metals, Wiley, Hoboken, NJ, 5th edn, 2009 Search PubMed.
- S. Ahrland, K. Dreisch, B. Norén and Å. Oskarsson, Mater. Chem. Phys., 1993, 35, 281–289 CrossRef CAS.
- T. K. Dargel, R. H. Hertwig and W. Koch, Mol. Phys., 1999, 96, 583–591 CrossRef CAS.
- H.-B. Yi, H. M. Lee and K. S. Kim, J. Chem. Theory Comput., 2009, 5, 1709–1717 CrossRef CAS.
- P. Pérez-Galán, N. Delpont, E. Herrero-Gómez, F. Maseras and A. M. Echavarren, Chem.–Eur. J., 2010, 16, 5324–5332 Search PubMed.
- V. Lavallo, G. D. Frey, S. Kousar, B. Donnadieu and G. Bertrand, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 13569–13573 CrossRef CAS.
- E. Herrero-Gómez, C. Nieto-Oberhuber, S. López, J. Benet-Buchholz and A. M. Echavarren, Angew. Chem., Int. Ed., 2006, 45, 5455–5459 CrossRef.
- S. Doherty, J. G. Knight, A. S. K. Hashmi, C. H. Smyth, N. A. B. Ward, K. J. Robson, S. Tweedley, R. W. Harrington and W. Clegg, Organometallics, 2010, 29, 4139–4147 CrossRef CAS.
- D. V. Partyka, T. J. Robilotto, M. Zeller, A. D. Hunter and T. G. Gray, Organometallics, 2008, 27, 28–32 CrossRef CAS.
- Q.-S. Li, C.-Q. Wan, R.-Y. Zou, F.-B. Xu, H.-B. Song, X.-J. Wan and Z.-Z. Zhang, Inorg. Chem., 2006, 45, 1888–1890 CrossRef CAS.
- R. M. Hanson, J. Appl. Crystallogr., 2010, 43, 1250–1260 CAS.
- K. Ozutsumi and T. Kawashima, Polyhedron, 1992, 11, 169–175 CrossRef CAS.
- S. V. Lindeman, R. Rathore and J. K. Kochi, Inorg. Chem., 2000, 39, 5707–5716 CrossRef CAS.
- C. A. Tolman, Chem. Rev., 1977, 77, 313–348 CrossRef CAS.
- J. Rajput, A. T. Hutton, J. R. Moss, H. Su and C. Imrie, J. Organomet. Chem., 2006, 691, 4573–4588 CrossRef CAS.
- R. Landis and J. Halpern, Organometallics, 1983, 2, 840–842 CrossRef.
- F. M. Dixon, M. S. Masar, P. E. Doan, J. R. Farrell, F. P. Arnold, C. A. Mirkin, C. D. Incarvito, L. N. Zakharov and A. L. Rheingold, Inorg. Chem., 2003, 42, 3245–3255 CrossRef CAS.
- H. Werner, G. Canepa, K. Ilg and J. Wolf, Organometallics, 2000, 19, 4756–4766 CrossRef CAS.
- J. A. Osborn and R. R. Schrock, J. Am. Chem. Soc., 1971, 93, 3089–3091 CrossRef CAS.
- T. Osswald, I. S. Mikhel, H. Rüegger, P. Butti and A. Mezzetti, Inorg. Chim. Acta, 2010, 363, 474–480 CrossRef CAS.
- W. D. Jones and L. Dong, J. Am. Chem. Soc., 1989, 111, 8722–8723 CrossRef CAS.
- A. Ceriotti, G. Ciani and A. Sironi, J. Organomet. Chem., 1983, 247, 345–350 CrossRef CAS.
- P. B. Hitchcock, M. McPartlin and R. Mason, J. Chem. Soc. D, 1969, 1367–1368 RSC.
- M. Aresta, E. Quaranta and A. Albinati, Organometallics, 1993, 12, 2032–2043 CrossRef CAS.
- Z. Zhou, G. Facey, B. R. James and H. Alper, Organometallics, 1996, 15, 2496–2503 CrossRef CAS.
- T. A. Albright, P. Hofmann, R. Hoffmann, C. P. Lillya and P. A. Dobosh, J. Am. Chem. Soc., 1983, 105, 3396–3411 CrossRef CAS.
- J. S. Siegel, K. K. Baldridge, A. Linden and R. Dorta, J. Am. Chem. Soc., 2006, 128, 10644–10645 CrossRef CAS.
- M. Nambu, D. L. Mohler, K. Hardcastle, K. K. Baldridge and J. S. Siegel, J. Am. Chem. Soc., 1993, 115, 6138–6142 CrossRef CAS.
- (a) H. Staudinger, Ber. Dtsch. Chem. Ges., 1905, 38, 1735–1739 CrossRef; (b) T. T. Tidwell, Angew. Chem., Int. Ed., 2005, 44, 5778–5785 CrossRef CAS.
- E. C. Taylor, A. McKillop and G. H. Hawks, Org. Synth., 1972, 52, 36 CAS.
- S. Gambarotta, S. Strologo, C. Floriani, A. Chiesi-Villa and C. Guastini, Inorg. Chem., 1985, 24, 654–660 CrossRef CAS.
- D. J. Mindiola and G. L. Hillhouse, J. Am. Chem. Soc., 2002, 124, 9976–9977 CrossRef CAS.
- L. Giannini, E. Solari, S. Dovesi, C. Floriani, N. Re, A. Chiesi-Villa and C. Rizzoli, J. Am. Chem. Soc., 1999, 121, 2784–2796 CrossRef CAS.
- (a) F. Basuli, J. C. Huffman and D. J. Mindiola, Inorg. Chem., 2003, 42, 8003–8010 CrossRef CAS; (b) F. Basuli, B. C. Bailey, L. A. Watson, J. Tomaszewski, J. C. Huffman and D. J. Mindiola, Organometallics, 2005, 24, 1886–1906 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC reference numbers 789234–789238 and 789473–789474. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c1sc00375e |
| This journal is © The Royal Society of Chemistry 2011 |