Glucuronide directed molecularly imprinted solid-phase extraction: isolation of testosterone glucuronide from its parent drug in urine†
Serena
Ambrosini
a,
Sudhirkumar
Shinde
b,
Ersilia
De Lorenzi
*a and
Borje
Sellergren
b
aDepartment of Drug Sciences, University of Pavia, Via Taramelli 12, 27100, Pavia, Italy. E-mail: ersidelo@unipv.it; Fax: +39 0382 422975; Tel: +39 0382 987747
bINFU, Faculty of Chemistry, Technical University of Dortmund, Otto Hahn Strasse 6, 44221, Dortmund, Germany. E-mail: B.Sellergren@infu.uni-dortmund.de; Fax: +49 231 7554234; Tel: +49 231 7554082
First published on 28th October 2011
Abstract
Two molecularly imprinted polymers (MIPs) that we recently described to be class-selective for glucuronides have been successfully exploited for the molecularly imprinted solid-phase extraction (MISPE) of testosterone glucuronide (TG) from its parent drug (T) in urine. Both sorbents targeted the glucuronate fragment but feature different functional groups for binding the carboxylate anion, MIP1, a neutral 1,3-diarylurea group, and MIP2, a cationic imidazolium functionality. MISPE-HPLC-UV methods developed using both sorbents allowed the extraction of TG from its parent compound in urine samples spiked at 150, 300 or 600 ng mL−1 for TG and at 50 ng mL−1 for T. By comparing the performance of the two sorbents it came out that MIP1 is a more suitable SPE packing than MIP2, since it isolated the glucuronide with a higher precision (RSD 2–5%, n = 3) and with an enhanced enrichment factor (EF = 4.2). On the basis of these results, the imprinted receptor MIP1 can be applied for the direct extraction of TG in doping and clinical analysis and to selectively capture any other relevant glucuronated metabolite avoiding tedious deconjugation steps prior to quantification.
Introduction
Testosterone (T) is among the substances most used by athletes within the anabolic-androgenic steroids family. These substances lead to increased strength and enhanced recovery; hence, their administration is prohibited.1–4 The majority of steroids is present in urine in their conjugated form as glucuronides, but sulfates have also been found.5,6 According to the World Anti-Doping Agency (WADA), a urine sample containing either a ratio of testosterone glucuronide to its isomer epitestosterone glucuronide (TG/EG) ≥ 4.0 or a concentration of TG (or EG) > 200 ng mL−1 is considered suspicious of testosterone abuse among males and requires further examination.4 The monitoring of TG or of other androgen glucuronides can also be used as a diagnostic tool, since steroid glucuronidation seems to be associated with increased risk for prostate cancer.7,8Nevertheless, the high hydrophilicity of glucuronated metabolites makes their analysis a difficult task. In doping control TG is still determined through indirect methods. The glucuronide has to be hydrolyzed to the corresponding free testosterone, which is subsequently isolated by liquid–liquid extraction (LLE), derivatized to form trimethylsilyl ethers and subjected to gas chromatography-mass spectrometry (GC-MS) analysis.3,9–13
The direct analysis of TG through the LC-MS technique after solid-phase extraction (SPE) is highly preferable, as it does not require the time-consuming and laborious steps of hydrolysis and derivatization that may produce inaccurate results. However, the main issue that must be overcome in the routine use of such methods is matrix effect. The literature available on the analysis of TG and steroid glucuronides gives evidence that signal suppression occurs when the glucuronated metabolite is not well separated from its parent drug or from other polar matrix interferents, thus limiting the precision, accuracy and sensitivity of the method.3,6,14,15 The key point and the present bottleneck to develop a LC-MS method for direct quantification of TG imply the use of an optimum sample preparation. To this regard, the application of molecular imprinted polymers (MIPs) to SPE—namely, molecularly imprinted solid-phase extraction (MISPE)—may offer a solution.
The synthesis of MIPs entails the polymerization of functional monomer(s) in the presence of a template molecule. Removal of the template molecule leaves behind cavities within the cross-linked polymer that are complementary to the original template and/or its analogous structures. These polymeric materials are intrinsically characterized by high levels of affinity and selectivity and hence they allow a customized sample pretreatment that minimizes background to be performed.16–20
One of the major limitations of MISPE is the poor recognition property of MIPs in aqueous media (e.g., biological samples). An additional step (LLE or SPE) before the selective MISPE protocol may need to be performed in order to transfer the analyte to an organic solvent where the MIP shows specific interactions for the target compound. Current trends are towards the development of water-compatible MIPs that allow the direct loading of aqueous samples.21–24 In this case, during the loading step the MIP behaves as a reversed-phase sorbent, capturing the analyte and matrix components by means of non-specific interactions. Then the percolation, prior to the elution step, of an optimized washing solvent discloses the imprinting effect and leads to the selective removal of matrix interferents.
We have recently described for the first time25 the synthesis of MIPs for the class-selective recognition of glucuronides. The MIPs were prepared by following the substructure approach along with the stoichiometric imprinting. The former was based on the use of glucuronic acid as template, preventively derivatized either to make it compatible with the organic solvents used during polymerization or to simulate any drug-like molecular structure linked to glucuronic acid. With the aim of improving the performance of MIPs in aqueous systems, either a urea- or a cationic imidazolium-based monomer that stoichiometrically interacts with the carboxylic function of the template was employed. Among the eleven MIPs produced, two have shown a high specificity for the glucuronated form of all the tested compounds—namely, testosterone, cotinine and mycophenolic acid. The diversity of the chemical structure of these compounds proves the class-selectivity against glucuronides of the new imprinted receptors.
As reported in this work, these two MIPs successfully accomplish the direct extraction of testosterone glucuronide from its parent drug in urine sample and importantly circumvent the cumbersome deconjugation step prior to analysis.26
The MISPE protocol was first optimized in artificial urine and then successfully implemented to real urine. Although the recovery of testosterone glucuronide was not quantitative, it was highly reproducible, leading in turn to a good analytical precision. Hence the MISPE protocol setup for the urea-based MIP1 has proven to be an efficacious sample clean-up procedure for the direct analysis of TG. Moreover, since glucuronidation is a common phase II reaction of human metabolism, this imprinted sorbent would positively impact all those areas of analysis involving glucuronide detection (e.g., clinical, environmental and forensic analysis).3,27–32
Experimental
Reagents
The templates 1,2,3,4-tetra-O-acetyl-β-glucuronic acid (TAG) and 1-O-dodecyl-β-glucuronic acid were synthesized according to the literature.33,34 1-O-Dodecyl-β-glucuronic acid sodium salt (DGNa+) was prepared by salification with 1 mM aqueous NaOH. The functional monomers 1-(4-vinylphenyl)-3-(3,5-bis(trifluromethyl)phenyl)urea (FM1) and 1-benzyl-3-vinyl-2,3-dihydro-1H-imidazolium bromide (FM2) were prepared as previously described by us.25,35 The structures of templates and monomers are shown in Fig. 1.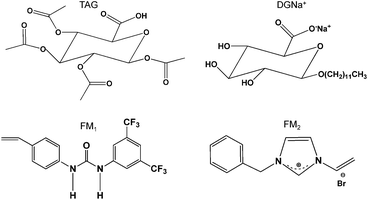 | ||
| Fig. 1 Chemical structure of the template TAG and DGNa+ and of the functional monomers FM1 and FM2. | ||
1,2,2,6,6-Pentamethylpiperidine (PMP), ethylene glycol dimethacrylate (EDMA), and pentaerythritol triacrylate (PETRA) were purchased from Sigma-Aldrich (Steinheim, Germany). The initiator 2,2′-azobis(2,4-dimethylvaleronitrile) (ABDV) was supplied from Wako (Neuss, Germany). All chemicals for the synthesis of the template, functional monomers and polymers were of reagent grade and used as received with the exception of EDMA. EDMA was washed consecutively with 10% NaOH, water, and brine; it was then dried over Na2SO4; and distilled under reduced pressure. All solvents used for synthesis were purchased from Sigma-Aldrich and purified according to the guidelines in Purification of Laboratory Chemicals. Anhydrous solvents for polymer preparation were stored over appropriate molecular sieves.
Testosterone (T) was bought from Sigma-Aldrich (Steinheim, Germany) whereas testosterone-β-D-glucuronide (TG) was purchased from Steraloid Inc. (Newport, USA). Chemical structures of T and TG are reported in Fig. 2. The salts NaN3, NaH2PO4 and Na2HPO4 were obtained by Merck (Darmstadt, Germany). NaCl and urea were purchased from BDH Laboratory Supplies (Poole, England). Methanol (MeOH), acetonitrile (MeCN), acetic acid (AcOH), HCOOH and H3PO4 for analysis were of HPLC grade and were supplied from Carlo Erba (Milan, Italy). LC-grade water was prepared using a Millipore Direct-QTM system (Bedford, MA, USA).
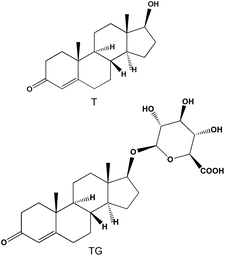 | ||
| Fig. 2 Chemical structures of T and TG. | ||
Polymer preparation
The two imprinted polymers MIP1 (template: TAG (181 mg, 0.5 mmol); base: PMP (78 mg, 0.5 mmol); functional monomer: FM1 (187 mg, 0.5 mmol); cross-linker: PETRA (3967 mg, 13.3 mmol); porogen: MeCN (5.6 mL)) and MIP2 (template: DGNa+ (96 mg, 0.25 mmol); functional monomer: FM2 (66 mg, 0.25 mmol); cross-linker: EDMA (1980 mg, 10 mmol); porogen: MeOH (2.8 mL)) were prepared by bulk polymerization as reported in our previous work.25 Briefly, template, functional monomer, the base PMP (if present in the polymer composition), cross-linker and the initiator ABDV (1% w/w of the total monomers) were dissolved with the appropriate dry porogen. The mixture was transferred into a polymerization tube and purged with N2 for 10 min. The polymerization tube was sealed and polymerization was thermally initiated by placing the tube in a water bath set at 45 °C for 24 h. After the end of polymerization, the tube was broken and the resultant solid polymer monolith was crushed into smaller fragments. The polymer fragments were then washed by soxhlet extraction for 24 h with MeOH![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :HCl 0.5 M (80:20, v/v) to remove the template, residual monomer(s) and initiator.
:HCl 0.5 M (80:20, v/v) to remove the template, residual monomer(s) and initiator.
The washed polymer fragments were crushed using a mortar and then sieved to obtain the desired particle size (36–50 μm). The polymers were sedimented several times using MeOH:H2O (80:20, v/v) to eliminate smaller particles. Control polymers (non-imprinted polymers, NIPs) were prepared in the absence of template and of the base when this was added to the pre-polymerization solution of the corresponding MIP.
Chromatographic apparatus
All chromatographic analyses were carried out on an Agilent HP1100 system (Agilent Technologies, Waldbronn, Germany), equipped with a multi-solvent delivery system, autosampler, on-line solvent degasser, column thermostat and DAD UV detector. The MISPE was carried out using empty SPE cartridges (1 mL) (Alltech, Sedriano, Italy), polyethylene frits (20 μm, Alltech, Sedriano, Italy) and a Baker SPE 12G vacuum unit.Preparation of spiked artificial urine
Stock solutions of TG (0.1 mg mL−1) and T (1 mg mL−1) in MeOH were separately prepared by dissolving an accurate amount of analyte in a 10 mL volumetric flask. The stock solutions were further diluted with MeOH to obtain working standard solutions of 10 μg mL−1 for each compound. Artificial urine was prepared by dissolving urea (20 g L−1) and NaCl (10 g L−1) in H2O and spiked with TG and T working standard solutions to give a final concentration of 300 ng mL−1 and 50 ng mL−1, respectively. NaN3 (0.1% w/v) was added in order to prevent any microbial growth and TG hydrolysis in aqueous reagents.36Optimized MISPE protocol in artificial urine
The empty cartridges were dry-packed manually by weighing 30 mg of each polymer. Frits were used (top and bottom) to prevent loss of the particles in the bed. The cartridges were conditioned with 2 × 1 mL of H2O and then loaded with 3 mL (for MIP1) or 0.4 mL (for MIP2) of spiked artificial urine. To selectively wash out the parent drug, 1 mL of MeCN:H2O:AcOH (97.99:2:0.01, v/v/v) or 4 × 1 mL of MeCN:H2O (15:85, v/v) was percolated through MIP1 or MIP2 cartridges, respectively. Finally, the glucuronide was eluted with 1 mL of MeCN:HCOOH (99:1, v/v) and the cartridges were re-equilibrated by percolating 1 mL of MeCN:HCOOH (99:1, v/v), to assure the elution of any residue of TG, followed by 2 × 1 mL of MeOH.
All loading fractions and washing fractions with an aqueous content higher than 50% were directly injected into the HPLC, whereas the washing fractions with an aqueous content lower than 50% and all the elution fractions were collected, evaporated under nitrogen, reconstituted with 250 μL of H2O, and finally injected (100 μL).
The specificity of the extraction was evaluated by applying the same MISPE procedure on the respective NIP cartridge. The enrichment factor (EF) was calculated as the ratio of analyte concentration in the reconstituted extract and that in the loaded sample.
Analysis of real urine
Urine was collected in the morning (10 a.m.). The measured pH was 7.09. After filtration on a cellulose membrane (0.45 μm pore size), NaN3 (0.1% w/v) was added.36 All urine samples were then stored at −20 °C until use. Subsequently, aliquots were spiked with the working standard solutions of TG and T, prepared in MeOH from the stock solutions, to give the desired concentration—namely, 150 ng mL−1 (low level), 300 ng mL−1 (medium level) and 600 ng mL−1 (high level) for TG and 50 ng mL−1 for T. Before MISPE each spiked urine sample was diluted with phosphate buffer (5 mM, pH = 7.0) (1:2, v/v) or H2O (1:1, v/v) when the extraction was performed on an MIP1 or MIP2 cartridge, respectively.
The MISPE protocol applied to real urine samples involved the same conditions as those optimized in artificial urine, except for the washing and/or the re-equilibration steps. In the case of MIP1 the washing volume was increased from 1 mL to 2 × 1 mL to obtain a cleaner extract. All packed cartridges were re-equilibrated by percolating 2 × 1 mL of MeCN:HCOOH (99:1, v/v) followed by 2 × 1 mL of MeOH, to guarantee the complete washing of the sorbent from any residue of the glucuronide and any other interferent present in the urine samples.
HPLC-UV analysis
The SPE fractions were analyzed on a column Hypersil ODS, 5 μm packing, 150 × 4.6 mm i.d. The elution of TG and T was performed using a flow rate of 1 mL min−1 and a mobile phase composed of 0.05% H3PO4 in MeCN (solvent A) and 0.05% H3PO4 (aqueous) (solvent B). The injection volume was 100 μL and the detector wavelength was set at 245 nm. The fractions collected from MISPE experiments in artificial urine samples were analysed keeping fixed the column temperature at 25 °C and using a gradient elution of 0–1 min A (10%), 1–11 min A (10–85%), 11–12 min A (85–90%), 12–13 min A (90%), 13–14 min A (90–10%), and 14–17 min A (10%). Unextracted artificial urine spiked with TG and T was directly injected into the HPLC as a reference mixture (intra-day injections) to establish the retention time of the analytes and to prove the peak identity. For quantification purposes of the fractions obtained from the MIP1 cartridge, an external linear calibration curve was built in the range between 15 ng mL−1 and 3600 ng mL−1 for TG, and between 15 ng mL−1 and 600 ng mL−1 for T. Taking into account the lower loading volume on MIP2, more narrow ranges were built to quantify the analytes collected from this cartridge, namely, between 15 ng mL−1 and 500 ng mL−1 for TG, and between 15 ng mL−1 and 90 ng mL−1 for T.The evaluation of the eluted fractions obtained from MISPE in real urine samples was carried out setting column temperature at 45 °C and employing a gradient program slightly different from that described above. In detail a gradient profile of 0–7 min A (10–50%), 7–11 min A (50%), 11–12 min A (50–90%), 12–15 min A (90%), 15–16 min A (90–10%), and 16–19 min A (10%) was used for fractions collected from the MIP1 cartridge, whilst a gradient elution of 0–7 min A (10–45%), 13–14 min A (45–90%), 14–17 min A (90%), 17–18 min A (90–10%), and 18–21 min A (10%) was applied for those obtained from the MIP2 cartridge. In order to quantify the extracts, blank urine samples were preconcentrated in both MIP cartridges following the optimized MISPE protocols, checked for not containing the analytes at the method detection limit and spiked with working standard solutions of TG and T. Linear calibration curves in the range between 15 ng mL−1 and 2400 ng mL−1 for TG and between 15 ng mL−1 and 200 ng mL−1 for T were built to quantify the extracts obtained from the MIP1 cartridge, whereas linear calibration ranges between 15 ng mL−1 and 500 ng mL−1 for TG and between 15 ng mL−1 and 90 ng mL−1 for T were built to calculate the recovery of the analytes after MISPE on MIP2. All calibration curve equations and correlation coefficients are reported in the ESI (Fig. S1†).
The limit of detection (LOD) and the limit of quantification (LOQ) were the minimum detectable amounts of analyte in blank urine extracted and then spiked giving a signal-to-noise ratio of 3 or 10, respectively. For both TG and T the LOQ was 15 ng mL−1, whereas the LOD was 10 ng mL−1.
The identity of glucuronide and the parent drug were confirmed by the UV spectrum and by comparing the chromatograms obtained after the injection of the extract of spiked urine with those obtained after the injection of the extracts of the blank urine and of the blank urine spiked only after performing the MISPE protocol (see ESI, Fig. S2 and S3†).
Results and discussion
Optimization of the SPE procedure in artificial urine
Urea and NaCl are the most abundant constituents of physiological human urine. Additionally, NaCl may greatly affect both recovery and repeatability of the MISPE procedure by increasing the counterion competition, especially when the cationic monomer FM2 is used. Given these premises, it was decided to first optimize the MISPE protocol in an aqueous solution of these components with the aim of simulating the matrix effect of real urine.As anticipated in the Introduction, a urinary concentration of TG > 200 ng mL−1 serves as a marker for possible testosterone abuse among males.4 The free steroid (T) occurs at extremely low levels in urine, given that less than 3% of testosterone is excreted as free fraction.37 Therefore, the MISPE experiments were carried out by using artificial urine spiked at 300 ng mL−1 and 50 ng mL−1 for TG and T, respectively. A TG concentration of 300 ng mL−1 was selected in order to reproduce altered profiles of TG in urine, whilst a T concentration of 50 ng mL−1 was chosen so as to avoid UV detection problems and to test the recognition properties of the MIP when the parent drug concentration is quite lower than that of the glucuronide.
The elution step was established in the first instance. With the use of an organic solvent in the presence of an acid, such as formic acid (1%), we expected to effectively disrupt the interactions between the analyte and the sorbent functional groups and we therefore tested mixtures of MeCN and formic acid as eluents. The application of 1 mL of MeCN:HCOOH (99:1, v/v) allowed the quantitative recovery of both T (MIP1: R 94%, RSD 6%, n = 2; MIP2: R 103%, RSD 6%, n = 2) and TG (MIP1: R 102%, RSD 6%, n = 2; MIP2: R 102%, RSD 11%, n = 2) from the two MIP cartridges. Hence, this elution solvent was selected for further experiments.
As described in our previous work,25MIP1 and MIP2 show an excellent specific recognition of glucuronide from its parent drug in MeCN:H2O:AcOH (97.99:2:0.01, v/v/v) or MeCN:H2O (30:70, v/v), respectively. Therefore these were the first solvent mixtures tested to selectively remove the parent compound T from the corresponding imprinted cartridges.
With regards to MIP1, 0.3 mL of MeCN:H2O:AcOH (97.99:2:0.01, v/v/v) was the minimum volume required to totally wash out T and extract the glucuronide (41%). The percolation of a higher washing volume, e.g. 1 mL, does not significantly affect TG recovery while it further reduces the non-specific interactions. This is clearly demonstrated by the extract obtained from NIP1, where TG recovery decreases from 20% to 7%. So as to reduce the loss of TG, 1 mL of MeCN with the addition of AcOH (0.01%) was tested. A higher glucuronide recovery was obtained exclusively from the non-imprinted cartridge (36%), thus giving evidence that the presence of 2% H2O is essential to disclose the imprinting effect. The hydrogen bond capability of H2O, added in such a small percentage, is probably enough to disrupt most non-specific binding effects, whilst specific ones are not influenced at all.38
As far as the optimization of the washing step on MIP2, the percolation of only 0.2 mL of MeCN:H2O (30:70 v/v) causes a dramatic loss of both glucuronide and T. Moreover, the latter is still detected in the elution fraction (data not shown). So as to minimize the loss of TG, it was decided to test sequential washings, 4 × 0.2 mL or 4 × 1 mL of MeCN:H2O (15:85, v/v). We anticipated that a higher water content in the washing fraction could strengthen the binding of both glucuronide and the parent drug to the polymer matrix due to hydrophobic effects. Eventually, a sequential washing of 4 × 1 mL allowed a complete removal of T and a good recovery of TG (62%) from the MIP2 cartridge. In contrast, NIP2 releases most of the glucuronide during the loading and washing steps resulting in a much lower recovery of TG (7%).
The final step of the MISPE optimization was to identify the highest sample volume which could be loaded on the SPE cartridges without significant breakthrough. On the MIP1 cartridge, TG starts to break through in the collected loading fractions after loading more than 3 mL of spiked artificial urine. In contrast, glucuronide breakthrough occurs on MIP2 by applying only 0.4 mL of spiked artificial urine. Based on these results, a loading volume of 3 mL or 0.4 mL was established for MIP1 and MIP2, respectively.
Analysis of artificial urine
Once the MISPE protocols had been established, analyses in triplicate of spiked artificial urine were performed on both MIP and NIP cartridges. The % recovery of TG in each fraction is summarized in Table 1 (the recoveries of T were below the LOD and are therefore not reported). It is evident that the recoveries of TG on the two MIPs are always higher than those obtained on the respective control sorbents. This proves that the capture of TG on the imprinted cartridge is achieved through selective and specific interactions.| MIP1 | NIP1 | MIP2 | NIP2 | |
|---|---|---|---|---|
| Replicate 1 | 32 | 9 | 62 | 7 |
| Replicate 2 | 35 | 6 | 58 | 6 |
| Replicate 3 | 37 | 9 | 60 | 39 |
| Mean | 35 | 8 | 60 | 17 |
| RSD % | 6 | 17 | 3 | 108 |
Despite low recovery, the high loading volume (3 mL) on MIP1 led to an enrichment factor for TG of 4.2, which is markedly higher than that achieved on MIP2 (EF = 1.0). This is also evident from the representative chromatograms reported in Fig. 3.
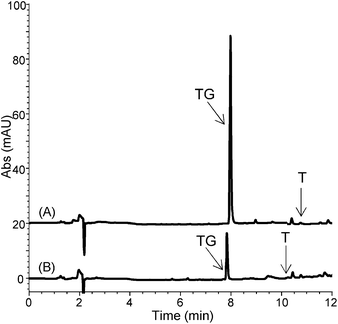 | ||
| Fig. 3 Chromatograms of spiked artificial urine after the optimized MISPE protocol on (A) MIP1 and (B) MIP2. | ||
From this series of experiments it can be concluded that MIP1 is better suited as an SPE sorbent for extraction of TG from real urine samples.
Analysis of real urine
The MIPs were then used to extract the glucuronide from its parent drug in real urine samples. As mentioned in the Experimental section, an extra dilution step of the urine matrix for both MIP cartridges and an additional washing step for MIP1 were necessary to reach a good compromise between recovery and sample clean-up. The recovery of the parent drug in the MIP extracts was low and below the LOD at each concentration level (n = 3) (see ESI, Fig. S4 and S5†). The results obtained for TG are summarized in Table 2.| Spiked level (TG–T, ng mL−1) | MIP1 | MIP2 |
|---|---|---|
| 600–50 | 32 (2) | 38 (10) |
| 300–50 | 28 (5) | 47 (21) |
| 150–50 | 34 (3) | 32 (19) |
By comparing the data achieved in artificial urine (Table 1), it is evident that accuracy and precision of the MISPE procedure on MIP2 drastically decrease when moving to a real urine sample, whereas they do not significantly vary for the MIP1 cartridge. Repeating the extraction procedure on the corresponding NIP cartridges led to significantly lower recoveries of both the parent drug (not detected) and the glucuronide (≤3%) (see ESI, Fig. S6 and S7†). This further confirms the specificity of the extraction provided by the MIP cartridges.
Stability and regeneration of the MISPE cartridges were not investigated. Nevertheless, the same polymer material was used throughout the studies—i.e., MIP and NIP used for the HPLC characterization25 were unpacked from the chromatographic columns and repacked into the SPE cartridges. This at least indicates that polymers, and MIP1 in particular, retain specificity and precision for a long-time usage under stressed conditions.
Conclusions
We conclude that the neutral urea-based MIP1 is a more suitable SPE sorbent compared to the cationic MIP2 for the selective enrichment of TG in urine sample. This is further corroborated by the chromatograms of the elution fractions (Fig. 4). Whereas the MIP2 extract appears unclean with interferents eluting close to TG, the corresponding MIP1 extract shows a more effective TG enrichment with less coextracted interferents. The lower selectivity of MIP2 is likely due to its cationic nature where coextraction of urine abundant anionic species occurs. This problem is overcome using the neutral MIP1 featuring only hydrogen bonding urea functional groups.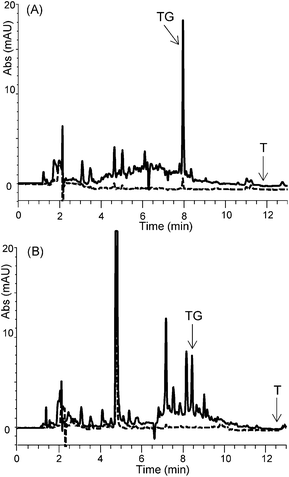 | ||
| Fig. 4 Chromatograms of spiked urine with TG (300 ng mL−1) and T (50 ng mL−1) after MISPE on (A) NIP1 and MIP1 and (B) NIP2 and MIP2 (NIP, dashed line; MIP, solid line). | ||
Although the recovery of testosterone glucuronide is low (28–34%), the MISPE protocol allows the direct determination of the conjugated metabolite with an excellent precision (RSD 2–5%, n = 3). This MIP recognizes the glucuronide moiety of the analyte, and therefore it would be useful to directly quantify not only testosterone glucuronide, but also those compounds which undergo the glucuronidation process, thus offering an important new tool for metabolite detection.
Notes and references
- A. T. Kicman, Br. J. Pharmacol., 2008, 154, 502 CrossRef CAS
.
- C. Saudan, N. Baume, N. Robinson, L. Avois, P. Mangin and M. Saugy, Br. J. Sports Med., 2006, 40, 21 CrossRef
- R. L. Gomes, W. Meredith, C. E. Snape and M. A. Sephton, Anal. Bioanal. Chem., 2009, 393, 453 CrossRef CAS
- WADA Technical Document—TD2009EAAS, Endogenous Anabolic Androgenic Steroids Testing, Reporting and Interpretive Guidance, 2009, http://www.wada-ama.org/Documents/News_Center/WADA_TD2009EAAS_Endogenous_Anabolic_Androgenic_Steroids_Oct2009.pdf Search PubMed.
- W. Schanzer, Clin. Chem., 1996, 42, 1001 CAS
- D. J. Borts and L. D. Bowers, J. Mass Spectrom., 2000, 35, 50 CrossRef CAS
- S. Chouinard, O. Barbier and A. Bélanger, J. Biol. Chem., 2007, 282, 33466 CrossRef CAS
- A. H. Karypidis, M. Olsson, S. O. Andersson, A. Rane and L. Ekström, Pharmacogenomics J., 2008, 8, 147 CrossRef CAS
- L. Dehennin, P. Lafarge, P. Dailly, D. Bailloux and J. P. Lafarge, J. Chromatogr., Biomed. Appl., 1996, 687, 85 CrossRef CAS
- E. Raynaud, M. Audran, J. C. Pagès, C. Fédou, J. F. Brun, J. L. Chana and A. Orsetti, Clin. Endocrinol., 1993, 38, 353 CrossRef CAS
- R. L. Gomes, W. Meredith, C. E. Snape and M. A. Sephton, J. Pharm. Biomed. Anal., 2009, 49, 1133 CrossRef CAS
- L. D. Bowers, Annu. Rev. Anal. Chem., 2009, 2, 485 CrossRef CAS
- C. Jiménez, R. de la Torre, J. Segura and R. Ventura, Rapid Commun. Mass Spectrom., 2006, 20, 858 CrossRef
- O. J. Pozo, K. Deventer, P. Van Eenoo and F. T. Delbeke, Rapid Commun. Mass Spectrom., 2007, 21, 2785 CrossRef CAS
- C. Saudan, J. M. Entenza, N. Baume, P. Mangin and M. Saugy, J. Chromatogr., B: Anal. Technol. Biomed. Life Sci., 2006, 844, 168 CrossRef CAS
- B. Sellergren, Anal. Chem., 1994, 66, 1578 CrossRef CAS
- Y. Xia, J. E. McGuffey, S. Bhattacharyya, B. Sellergren, E. Yilmaz, L. Wang and J. T. Bernert, Anal. Chem., 2005, 77, 7639 CrossRef CAS
- M. M. Ariffin, E. I. Miller, P. A. G. Cormack and R. A. Anderson, Anal. Chem., 2007, 79, 256 CrossRef CAS
- J. Haginaka, J. Sep. Sci., 2009, 32, 1548 CrossRef CAS
- E. Turiel and A. Martín-Esteban, Anal. Chim. Acta, 2010, 668, 87 CrossRef CAS
- B. Dirion, Z. Cobb, E. Schillinger, L. I. Andersson and B. Sellergren, J. Am. Chem. Soc., 2003, 125, 15101 CrossRef CAS
- J. L. Urraca, M. C. Moreno-Bondi, A. J. Hall and B. Sellergren, Anal. Chem., 2007, 79, 695 CrossRef CAS
- E. Benito-Peña, J. L. Urraca, B. Sellergren and M. C. Moreno-Bondi, J. Chromatogr., A, 2008, 1208, 62 CrossRef
- E. Benito-Peña, S. Martins, G. Orellana and M. C. Moreno-Bondi, Anal. Bioanal. Chem., 2009, 393, 235 CrossRef
- S. Ambrosini, M. Serra, S. Shinde, B. Sellergren and E. De Lorenzi, J. Chromatogr., A, 2011, 1218, 6961 CrossRef CAS
- B. Tse Sum Bui, F. Merlier and K. Haupt, Anal. Chem., 2010, 82, 4420 CrossRef
- M. Shipkova and E. Wieland, Clin. Chim. Acta, 2005, 358, 2 CrossRef CAS
- I. Stepanov and S. S. Hecht, Cancer Epidemiol., Biomarkers Prev., 2005, 14, 885 CAS
- W. Dekant and W. Völkel, Toxicol. Appl. Pharmacol., 2008, 228, 114 CrossRef CAS
- T. Kraemer and L. D. Paul, Anal. Bioanal. Chem., 2007, 388, 1415 CrossRef CAS
- F. Musshoff, J. Trafkowski, U. Kuepper and B. Madea, J. Mass Spectrom., 2006, 41, 633 CrossRef CAS
- E. Zuccato, S. Castiglioni, R. Bagnati, C. Chiabrando, P. Grassi and R. Fanelli, Water Res., 2008, 42, 961 CrossRef CAS
- M. Tosin and P. V. Murphy, Org. Lett., 2002, 4, 3675 CrossRef CAS
- G. Milkereit, M. Morr, J. Thiem and V. Vill, Chem. Phys. Lipids, 2004, 127, 47 CrossRef CAS
- A. J. Hall, P. Manesiotis, M. Emgenbroich, M. Quaglia, E. De Lorenzi and B. Sellergren, J. Org. Chem., 2005, 70, 1732 CrossRef CAS
- M. Tsivou, D. Livadara, D. G. Georgakopoulos, M. A. Koupparis, J. Atta-Politou and C. G. Georgakopoulos, Anal. Biochem., 2009, 388, 179 CrossRef CAS
- J. J. Schulze, J. Lundmark, M. Garle, I. Skilving, L. Ekström and A. Rane, J. Clin. Endocrinol. Metab., 2008, 93, 2500 CrossRef CAS
- J. G. Karlsson, B. Karlsson, L. I. Andersson and I. A. Nicholls, Analyst, 2004, 129, 456 RSC
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c1an15606c |
| This journal is © The Royal Society of Chemistry 2012 |