Use of micellar mobile phases for the chromatographic determination of melamine in dietetic supplements
Beatriz
Beltrán-Martinavarro
,
Juan
Peris-Vicente
,
Sergio
Marco-Peiró
,
Josep
Esteve-Romero
,
Maria
Rambla-Alegre
and
Samuel
Carda-Broch
*
Química Bioanalítica, QFA, ESTCE, Universitat Jaume I, 12071, Castelló, Spain. E-mail: scarda@qfa.uji.es
First published on 7th November 2011
Abstract
Melamine is a nitrogen-rich industrial chemical which is occasionally used to increase the apparent protein content of different products destined for human and animal consumption. In this work, a liquid chromatographic procedure that uses micellar mobile phases of sodium dodecyl sulfate (SDS) buffered at pH 3, a C18 column and UV detection is reported for the determination of melamine in dietetic supplements. Samples were reconstituted with a SDS solution and were directly injected, thus avoiding long extraction and experimental procedures. Melamine was eluted in less than 10 min with no interference by other compounds of the matrices. The optimum mobile phase composition was taken by a chemometrical approach that considers the retention factor, efficiency and peak shape. Validation was performed following the indications of the European Commission (Decision 2002/657/EC). The following parameters were considered: linearity (0.02–100 μg mL−1; R2 = 0.9996), intra- and inter-day precisions (<12.4%), accuracy (90.0–101.3%), and robustness (less than 9.8% and 5.1%, for retention time and peak area, respectively). The limits of detection and quantification were 9 and 20 ng mL−1, respectively. Recoveries for several spiked samples were in the 85.8–114.3% range. These results indicate that the proposed methodology is useful for routine analysis of control quality of infant formula and adult dietetic supplements.
1. Introduction
Melamine, or 1,3,5-triazine-2,4,6-triamine (Fig. 1), is an organic compound commonly used in the synthesis of melamine formaldehyde resins for the manufacture of laminates, plastics and glues or adhesives. Melamine can be found at μg mL−1 levels in food and beverages due to migration from melamine-containing resins1 or as a metabolite product of cyromazine, an insecticide used on animals and crops.2 When deliberately added to a number of different types of materials in both animal and human food, it can increase the nitrogen concentration, which suggests a false increase in the protein concentration measured by the Kjeldahl method.3 | ||
| Fig. 1 Melamine structure. | ||
Dietetic supplements are a complex matrix due to the presence of many compounds destined to correct deficiencies in both adult and infant food. In March 2007, a pet food manufacturer alerted the US Food and Drug Administration (US FDA) to animal deaths in the United States which appeared to be linked to certain batches of pet food. Further investigation showed that raw materials (wheat gluten and rice protein), which had been imported from China and used to manufacture pet food,4 were apparently contaminated with melamine intentionally to increase their total nitrogen concentration and, consequently, the calculated protein content. In September 2008, melamine-tainted milk resulted in nephrolithiasis and renal failure in infants in China. More than 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 infants were hospitalised and six infant deaths were confirmed.5Melamine could form crystals in combination with cyanuric acid. Very recent Chinese data on the composition of renal calculi (stones) from 15 infants in China who had consumed contaminated infant formula (Chinese Center for Disease Control and Prevention, unpublished data, 2008) confirmed that these stones were composed of uric acid and melamine (in a 1.2:1 to 2.1:1 molar ratio), and no cyanuric acid was detected.6
000 infants were hospitalised and six infant deaths were confirmed.5Melamine could form crystals in combination with cyanuric acid. Very recent Chinese data on the composition of renal calculi (stones) from 15 infants in China who had consumed contaminated infant formula (Chinese Center for Disease Control and Prevention, unpublished data, 2008) confirmed that these stones were composed of uric acid and melamine (in a 1.2:1 to 2.1:1 molar ratio), and no cyanuric acid was detected.6
Due to the proved toxicity of melamine, the safety limit of melamine ingestion has been officially set by the US FDA at 2.5 μg mL−1 for adult's food,7,8 and at 1 μg mL−1 for infant formula.7 Therefore, a reliable method is needed to determine melamine residues in food, particularly in products for children, in order to eliminate the potential threat to human health.
Different methods based on capillary zone electrophoresis with diode array detection,9gas chromatography-tandem mass spectrometry,10,11high performance liquid chromatography with UV11,12 or mass spectrometry detection2,11,13,14 have been developed for the quantification of melamine in a large number of different food and agricultural matrices such as feeds,11 milk and milk products,9–11 dairy products and fish feed,9 cereal flours,13 chard,2 animal feed14 and royal jelly.12 However, due to the complexity of the matrices, these analytical methodologies involve time-consuming extraction, preconcentrations and purification steps such as molecular imprinting and solid-phase,11 solid–liquid2,12–14 and liquid–liquid9extraction. Moreover, because of the need for high selectivity, mobile phases are programmed as gradients, which make the analysis of a large amount of samples a difficult task, due to the time of stabilization of the column at the end of each analysis.
The use of surfactant-mediated mobile phases15 usually simplifies the experimental procedures that involve biological matrices, since their hydrophobic compounds (mainly proteins) are solubilised and become harmless for the chromatographic system. Moreover they are eluted with, or shortly after, the solvent front, and do not interfere with the analytes.16,17 The introduction of micellar media in the mobile phase and the consequent modification of the stationary phase increase the number of interactions inside the column (between stationary phase, mobile phase, micelles and analyte). A chemometrics approach provides a simulation of the elution conditions, from the data obtained by the study of the analytes in few several mobile phases following an experimental design.18,19
In a previous work, the authors analysed melamine in milk samples using a hybrid micellar mobile phase of sodium dodecyl sulfate and propanol.20 Nowadays, it is desirable to work with non-pollutant mobile phases, which means free of organic solvents or containing low amounts of them. The aim of this work is to perform an easy, fast, accurate and reliable analytical methodology to quantify the level of melamine in dietetic supplements using pure micellar mobile phases containing only the anionic surfactant sodium dodecyl sulfate in the absence of any organic solvent. The analyte has to be resolved from the other compounds of the matrix with sufficient sensitivity to reach the safety levels marked by the FDA. The proposed method was validated following EC indications in terms of selectivity, linearity, sensitivity, repeatability, intermediate precision, robustness and recovery.21 Since safety limits are not indicated by EU regulations, those established by the US FDA have been considered.7,8
2. Materials and methods
2.1. Chemicals and reagents
Melamine (>99% purity) was purchased from Aldrich (St Louis, MO, USA). Sodium dodecyl sulfate (SDS, >99% purity) was from Merck (Darmstadt, Germany). Ultrapure water (Millipore S.A.S., Molsheim, France) was used throughout to prepare the mobile phases. Sodium dihydrogenophosphate monohydrate and HCl were obtained from Panreac (Barcelona, Spain). Methanol was bought from J. T. Baker (Deventer, The Netherlands) and 1-propanol and 1-butanol obtained from Scharlab (Barcelona). The characteristics of the studied dietetic supplements are shown in Table 1 (they were all purchased in a local pharmacy).| Commercial name | Supplier | State | Consumer | Spiked conc. /μg mL−1 | Recovery (%) (n = 6) |
|---|---|---|---|---|---|
| PediaSure Vanilla | Abbot Laboratories Spain (Madrid, Spain) | Powder | Infants | 0.050 | 97.8 |
| 0.100 | 87.8 | ||||
| 0.150 | 94.4 | ||||
| 5 | 114.0 | ||||
| PediaSure Drink Vanilla | Liquid | Infants | 0.050 | 90.9 | |
| 0.100 | 91.9 | ||||
| 0.150 | 102.1 | ||||
| 5 | 114.0 | ||||
| PediaSure Chocolate | Powder | Infants | 0.050 | 93.4 | |
| 0.100 | 112.3 | ||||
| 0.150 | 88.5 | ||||
| 5 | 109.4 | ||||
| PediaSure Drink Chocolate | Liquid | Infants | 0.050 | 85.3 | |
| 0.100 | 86.2 | ||||
| 0.150 | 90.5 | ||||
| 5 | 111.9 | ||||
| Nutrinovex Complex Chocolate | Grupo Farmacéutico de Levante, (Castelló, Spain) | Powder | Adults | 0.125 | 114.1 |
| 0.250 | 101.6 | ||||
| 0.375 | 87.9 | ||||
| 5 | 114.3 | ||||
| Nutrinovex Whey Chocolate | Powder | Adults | 0.125 | 90.4 | |
| 0.250 | 107.6 | ||||
| 0.375 | 92.5 | ||||
| 5 | 112.3 | ||||
| Nutrinovex Endurance | Powder | Adults | 0.125 | 87.9 | |
| 0.250 | 113.8 | ||||
| 0.375 | 100.0 | ||||
| 5 | 111.8 | ||||
| Nutrinovex Complex Strawberry | Powder | Adults | 0.125 | 106.7 | |
| 0.250 | 85.8 | ||||
| 0.375 | 87.6 | ||||
| 5 | 102.5 | ||||
| Optifast Coffee | Nestle Spain, (Espulgues de Llobregat, Spain) | Powder | Adults | 0.125 | 89.7 |
| 0.250 | 113.9 | ||||
| 0.375 | 96.4 | ||||
| 5 | 95.9 | ||||
| Optifast Strawberry | Powder | Adults | 0.125 | 107.0 | |
| 0.250 | 113.5 | ||||
| 0.375 | 102.1 | ||||
| 5 | 110.6 | ||||
| Meritene Chocolate | Powder | Adults | 0.125 | 105.0 | |
| 0.250 | 95.2 | ||||
| 0.375 | 87.5 | ||||
| 5 | 101.2 | ||||
| Meritene Junior Chocolate | Powder | Infants | 0.050 | 112.0 | |
| 0.100 | 105.2 | ||||
| 0.150 | 87.2 | ||||
| 5 | 94.5 |
2.2. Instrumentation
The chromatographic system, an Agilent Technologies series 1100 CA, USA, was equipped with an isocratic pump, a degasifier, an autosampler and a DAD (range 190–700 nm). The column used was a Kromasil C18 (150 mm × 4.6 mm i.d., pore size 100 Å and 5 μm particle size) from Scharlab. The pH of solutions was measured with a potentiometer model GLP 22 Crison (Barcelona), equipped with a combined Ag/AgCl/glass electrode. The analytical balance used was an AX105 Delta-Range (Metter-Toledo, Switzerland). The mobile phases and the injected solutions were filtered through 0.45 μm nylon membranes (Micron Separations, MA, USA). An ultrasonic bath was used to dissolve the standards (model Ultrasons-H, Selecta, Barcelona).2.3. Mobile phase, sample and standard preparation
The micellar mobile phases were prepared by dissolving the appropriate amount of SDS and dihydrogenophosphate monohydrate in ultrapure water, and then the pH was adjusted to the desired value. Finally, the solution was topped up with ultrapure water to the mark on the volumetric flask, sonicated and filtered.Samples were spiked by adding the appropriate amount of melamine to 1 mL of liquid dietetic supplement, and by filling up to 10 mL with a solution of 0.05 M SDS at pH 3. Powdered samples were reconstituted by dissolving 1 g in 10 mL of the same micellar solution.
Stock solutions with 1, 100, and 200 μg mL−1 of melamine were prepared by dissolving the appropriate amount in methanol.
2.4. Chromatographic conditions
Several mobile phases were tested by varying the SDS concentration, while the pH was buffered at 3. Separation was performed by running the mobile phase at 1 mL min−1 through a Kromasil C18 column thermostatted at 25 °C. The injection volume was 20 μL and the UV wavelength was set at 210 nm. The optimal mobile phase was finally an aqueous solution (0.15 M SDS, pH 3) without any organic solvent. Chromatographic signals were acquired with an Agilent ChemStation (Rev. A.10.01). Michrom software was used for processing the chromatographic data.192.5. Method validation
Method validation was performed following the criteria specified by the European Commission Decision 2002/657/EC (2002),21 and using spiked dietetic supplements, all of which provided similar results.Linearity and sensitivity were obtained by injecting the analyte at 15 different concentration levels to cover the whole working range (0.02–100 μg mL−1). Calibration curves of the spiked dietetic supplement samples were calculated by plotting the peak area of melamineversusmelamine concentration using the least squares linear regression analysis method. The limit of detection (LOD) was based on the 3s criterion (3s/b, three times the standard deviation of the lowest concentration solution included in the calibration divided by the slope of the calibration curve using a series of 10 solutions containing a low melamine concentration), and the limit of quantification (LOQ) was selected as the low concentration used in the calibration curve.
Decision limits (CCα) and detection capabilities (CCβ) were also calculated. CCα was calculated by analysing 20 samples spiked with melamine at the LOQ and safety limits (1 μg mL−1 for infants and 2.5 μg mL−1 for adults) of melamine. CCα was calculated as the concentration spiked plus 1.64 of the corresponding standard deviation. To obtain the CCβ values, 20 samples were spiked at the calculated CCα levels, and were analysed. The CCβ was calculated as the theoretical value of CCα previously obtained plus 1.64 times the standard deviation.21
Accuracy and precision were also determined by analysing three different concentration levels corresponding to 0.5, 1 and 1.5 times the proposed safety limits. Method robustness was also evaluated.
3. Results and discussion
3.1. Mobile phase selection and chromatographic optimization
Melamine is a polar compound (log Po/w = −1.14, pKa = 5.10), which means that adequate analysis times will be obtained using a C18 column and a pure micellar mobile phase. Mobile phases containing 0.05 and 0.1 M SDS were tested, but the retention time was too long. At 0.15 M SDS, the melamine peak eluted at 8.3 min, and was sufficiently separated from the matrix interferences.The addition of a short-chain alcohol (1-propanol and 1-butanol) can be used in micellar mobile phases to improve efficiency and to reduce the retention time of the compounds.22 However, in this case, the addition of 1-butanol produces a too fast elution (near the dead volume), and 1-propanol does not provide any significant advantage. Therefore, no alcohol was added to the mobile phase.
An interpretive optimisation strategy was followed to select the best surfactant concentration for the determination of melamine in dietetic supplements. Thus, several mobile phases containing the following SDS (M) concentrations were assayed: 0.05, 0.1, 0.15 and 0.2 M, all buffered at pH 3. This concentration range was selected to avoid excessive retention times or an elution near the void volume. Retention factors (k), efficiencies (N) and asymmetries (B/A) were measured for melamine, as well as for the front of the chromatogram and two unknown matrix compounds, which could overlap with the analyte. These data were processed with the Michrom software.19 The retention of the compounds was modelled according to:15
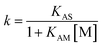 | (1) |
Fig. 2 depicts the resolution diagram. As can be observed, the resolution is poor below a concentration of 0.10 M SDS and above 0.16 M SDS due to several overlapping peaks. Although the best resolution was found at 0.14 M (R = 0.9997), the mobile phase selected was 0.15 M SDS, because the resolution was similar, and the retention time of melamine was significantly reduced. Melamine was adequately resolved from the other matrix peaks in less than 10 min using this mobile phase. The chromatographic parameters for melamine were: retention time, tR = 8.3 min, retention factor, k = 8.1, efficiency, N = 2450, and asymmetry factor, B/A = 1.1.
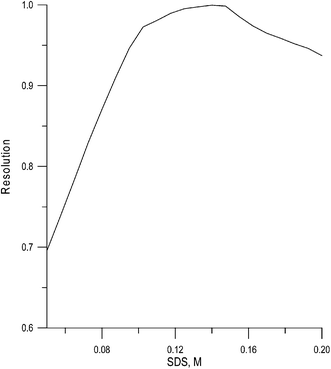 | ||
| Fig. 2 Contour map of global resolution for the separation of melamine in dietetic supplements. | ||
3.2. Method validation
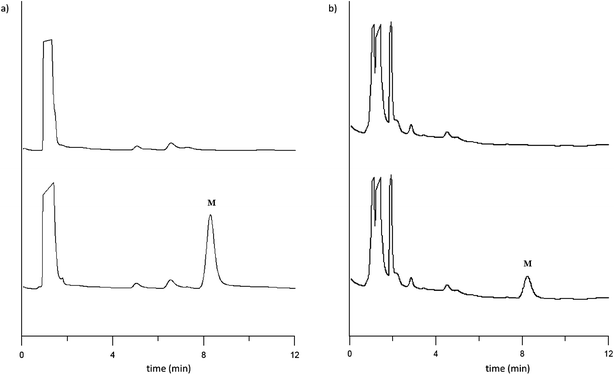 | ||
| Fig. 3 Chromatograms of: (a) blank (top) and spiked PediaSure Vanilla powder (infants) (bottom) at 5 μg mL−1, and (b) blank (top) and spiked Nutriben endurance (adults) (bottom) at 25 μg mL−1. Peak identification: M = melamine. Extracts were analysed following the optimised condition methodology. | ||
The slope and intercept were determined using the least squares linear regression analysis method. The adjusted equation and regression coefficient (R2), taken as the average of the five calibration curves, were:
| Peak area = (4.03 ± 0.06)[melamine] − (0.000 ± 0.003), R2 = 0.9996 |
The LOQ was the lower level reached in the calibration (20 ng mL−1), and the LOD was set at 9 ng mL−1 by the 3s criterion.
| Added concentration/μg mL−1 | Founda (mean ± SD)/μg mL−1 | Accuracy (%) | Intra-day RSD (%) | Foundb (mean ± SD)/μg mL−1 | Accuracy (%) | Inter-day RSD (%) |
|---|---|---|---|---|---|---|
| a n = 6. b n = 5. | ||||||
| 0.05 | 0.045 ± 0.005 | 90.0 | 11.1 | 0.050 ± 0.004 | 100 | 8.0 |
| 0.1 | 0.097 ± 0.012 | 97.0 | 12.4 | 0.101 ± 0.007 | 101 | 6.9 |
| 0.15 | 0.138 ± 0.006 | 92.0 | 4.4 | 0.148 ± 0.007 | 98.7 | 4.7 |
| 0.125 | 0.116 ± 0.007 | 92.8 | 6.0 | 0.120 ± 0.012 | 96.0 | 10.0 |
| 0.25 | 0.25 ± 0.02 | 100.0 | 8.0 | 0.25 ± 0.02 | 100 | 8.0 |
| 0.375 | 0.38 ± 0.03 | 101.3 | 7.9 | 0.38 ± 0.04 | 101.3 | 10.5 |
| 2 | 1.92 ± 0.02 | 96.0 | 1.0 | 1.90 ± 0.09 | 95.0 | 4.7 |
| 5 | 5.04 ± 0.07 | 101 | 1.4 | 4.8 ± 0.4 | 96.0 | 8.3 |
| 10 | 9.73 ± 0.04 | 97.3 | 0.4 | 9.5 ± 0.2 | 95.0 | 2.1 |
The intra-day analysis was determined by injecting the aliquots of these samples six times on the same day. Suitable precision (RSD < 12.4%) and accuracy (90.0–101.3%) were found. The inter-day analyses correspond to the average of five measurements of the intra-day values taken over a 3 month period. Precision, expressed as method repeatability, and accuracy were estimated from the recovery experiments (n = 5) at each concentration level. Excellent precision (RSD < 10.5%) and accuracy (95.0–101.3%) were obtained for all the matrices. The results indicate that the proposed methodology is suitable for the routine analysis of dietetic supplements.
| Chromatographic parameters | Level | Retention time/min | Area (arbitrary unit) |
|---|---|---|---|
| Concentration SDS (M) | |||
| 0.145 | −0.005 | 8.33 | 3.85 |
| 0.150 | 0 | 8.73 | 3.80 |
| 0.155 | +0.005 | 7.93 | 3.98 |
| Mean ± SD | 8.3 ± 0.4 | 3.88 ± 0.09 | |
| RSD (%) | 4.8 | 2.3 | |
| pH | |||
| 2.9 | −0.1 | 7.9 | 3.95 |
| 3.0 | 0 | 8.73 | 3.80 |
| 3.1 | +0.1 | 8.23 | 4.20 |
| Mean ± SD | 8.3 ± 0.6 | 4.0 ± 0.2 | |
| RSD (%) | 7.2 | 5.0 | |
| Flow/mL min −1 | |||
| 0.9 | −0.1 | 9.37 | 3.62 |
| 1.0 | 0 | 8.73 | 3.80 |
| 1.1 | +0.1 | 7.70 | 3.87 |
| Mean ± SD | 8.6 ± 0.8 | 3.76 ± 0.12 | |
| RSD (%) | 9.3 | 3.2 | |
In this case, three sets of CCα and CCβ were calculated, and the LOQ and the safety limits for infant formula and adult dietetic supplements were taken as the established limit.25 The obtained results were: spiking 20 ng mL−1 (LOQ level): CCα = 21 ng mL−1 and CCβ = 25 ng mL−1; spiking 0.1 μg mL−1 (safety limit for infants): CCα = 0.120 μg mL−1 and CCβ = 0.140 μg mL−1; and spiking 0.25 μg mL−1 (safety limit for adults): CCα = 0.280 μg mL−1 and CCβ = 0.210 μg mL−1. The obtained values indicate that melamine can be detected in contaminated samples with the established limits.
4. Conclusions
Micellar liquid chromatography has proved to be a fast, sensitive, and selective technique for the determination of melamine in a wide variety of dietetic supplements for adults and infant formula. The developed method allows the rapid determination of melamine by the direct injection of the reconstituted samples into the chromatographic system, after filtration, thus avoiding long, tedious extractions. The analysis time was under 10 min. Validation was performed according to EC guidelines with satisfactory results in the linearity, selectivity, accuracy, robustness and recovery studies. This method meets the requirements of the “green chemistry” concept since no organic solvent has been used. Besides, it is relatively inexpensive compared to other methods, thus making it a more appealing method. The proposed technique could be recommended as a routine method for the analysis of melamine in dietetic supplements for adults and infant formula.Acknowledgements
This work has been supported by a project from the Spanish Ministry of Education and Science (MEC) CTQ 2007 764473/BQU and the Foundation Caixa-Castelló, Bancaixa P1-1B2006-12. María Rambla-Alegre also wishes to acknowledge the MEC for the FPU grant.References
- H. Ishiwata, T. Inoue, T. Yamazaki and K. Yoshihira, J. Assoc. Off. Anal. Chem., 1987, 70, 457–460 CAS
.
- J. V. Sancho, M. Ibañez, S. Grimalt, O. J. Pozo and F. Hernandez, Anal. Chim. Acta, 2005, 530, 237–243 CrossRef CAS
- P. G. Wiles, I. K. Gray and R. C. Kissling, J. AOAC Int., 1998, 81, 620–632 CAS
-
FDA, Interim Melamine and Analogues Swafty/Risk Assessement, May 25 2007 Search PubMed
-
Times on line (From the Times), Chinese Milk Powder Contaminated with Melamine Sickens 1253 Babies, http://wwwtimesonline.co.uk/tol/news/world/asia/article4758549.ece, September 16 2008 Search PubMed
-
Report of a WHO Expert Meeting in Collaboration with FAO Supported by Health Canada, Otawa, 1–4 December 2008 Search PubMed
- L. Zhu, G. Gamez, H. Chen, K. Chingin and R. Zenobi, Chem. Commun., 2009, 559–561 RSC
- http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/2008/ucm116960.htm .
- N. Yan, L. Zhou, Z. Zhu and X. Chen, J. Agric. Food Chem., 2009, 57, 807–811 CrossRef CAS
- H. Miao, S. Fan, Y. N. Wu, L. Zhang, P. P. Zhou, J. G. LI, H. J. Chen and Y. F. Zhao, Biomed. Environ. Sci., 2009, 22, 87–94 CrossRef CAS
- L. He, Y. Su, Y. Zheng, X. Huang, L. Wu, Y. Liu, Z. Zeng and Z. Chen, J. Chromatogr., A, 2009, 1216, 6196–6203 CrossRef CAS
- J. Zhou, J. Zhao, X. Xue, F. Chen, J. Zhang, Y. Li, L. Wu and L. Chen, J. Sep. Sci., 2010, 33, 167–173 CrossRef CAS
- S. Ehling, S. Tefera and I. P. Ho, Food Addit. Contam., 2007, 24, 1319–1325 CrossRef CAS
- D. N. Heller and C. B. Nochetto, Rapid Commun. Mass Spectrom., 2008, 22, 3624–3632 CrossRef CAS
-
A. Berthod and M. C. García-Álvarez-Coque, Micellar Liquid Chromatography, Marcel-Dekker, New York, USA, 2000 Search PubMed
- J. Esteve-Romero, E. Ochoa-Aranda, D. Bose, M. Rambla-Alegre, J. Peris-Vicente and A. Martinavarro-Domínguez, Anal. Bioanal. Chem., 2010, 397, 1557–1561 CrossRef CAS
- M. Rambla-Alegre, J. Esteve-Romero and S. Carda-Broch, Anal. Chim. Acta, 2009, 633, 250–256 CrossRef CAS
- M. Rambla-Alegre, J. Peris-Vicente, J. Esteve-Romero and S. Carda-Broch, Food Chem., 2010, 123, 1294–1302 CrossRef CAS
-
J. R. Torres-Lapasió, Michrom Software, Marcel-Dekker, New York, USA, 2000 Search PubMed
- M. Rambla-Alegre, J. Peris-Vicente, S. Marco-Peiró, B. Beltrán-Martinavarro and J. Esteve-Romero, Talanta, 2010, 81, 894–900 CrossRef CAS
- 2002/657/EC, Commission decision of 12 August 2002 implementing council directive 96/23/EC concerning the performance of analytical methods and the interpretation of results, Off. J. Eur. Communities: Legis., 2002, 221, 8–62 Search PubMed
- M. Rambla-Alegre, M. T. Gil-Agustí, M. E. Capella-Peiró, S. Carda-Broch and J. Esteve-Romero, J. Chromatogr., B: Anal. Technol. Biomed. Life Sci., 2006, 839, 89–94 CrossRef CAS
- J. R. Torres-Lapasió, J. J. Baeza-Baeza and M. C. García-Álvarez-Coque, Anal. Chem., 1997, 69, 3822–3831 CrossRef
- E. Verdon, D. Hurtaud-Pessel and P. Sanders, Accredit. Qual. Assur., 2006, 11, 58–62 CrossRef CAS
-
K. Verschueren, Handbook of Environmental Data on Organic Chemicals, Van Nostrand Reinhold Co., New York, 3rd edn, 1996 Search PubMed
| This journal is © The Royal Society of Chemistry 2012 |