Target and non-target screening strategies for organic contaminants, residues and illicit substances in food, environmental and human biological samples by UHPLC-QTOF-MS†
Ramon
Díaz
,
María
Ibáñez
,
Juan V.
Sancho
and
Félix
Hernández
*
Research Institute for Pesticide and Water, University Jaume I, Av. Sos Baynat S/N, 12071, Castellón, Spain. E-mail: felix.hernandez@qfa.uji.es; Fax: +34 964387368; Tel: +34 964387366
First published on 28th November 2011
Abstract
In this paper, we illustrate the potential of ultra-high performance liquid chromatography (UHPLC) coupled with hybrid quadrupole time-of-flight mass spectrometry (QTOF MS) for large scale screening of organic contaminants in different types of samples. Thanks to the full-spectrum acquisition at satisfactory sensitivity, it is feasible to apply both (post)-target and non-target approaches for the rapid qualitative screening of organic pollutants in food, biological and environmental samples. Different strategies have been applied and compared in this work. The first approach consists of target screening based on automatically extracting the exact analyte masses with a narrow mass window (±10 mDa). The selection of analytes can be made after MS acquisition as non-specific analyte information is required when injecting the samples. The second, non-targeted approach, consists of a first component detection step followed by the search of the detected components in home-made spectral libraries. In this work, two types of libraries have been evaluated: a theoretical database, including the molecular formula of a large number of pollutants (∼1000), and an empirical mass spectra library which includes a lower number of compounds for which reference standards were available. In all cases the confidence of the identification process was excellent, thanks to the value of information given in QTOF MSE acquisition mode (i.e. simultaneous acquisition of low and high energy TOF MS spectra in a unique run). Both, target and non-target approaches, are complementary and both have advantages and drawbacks. Their application to different types of samples has allowed the detection of diverse organic compounds, for example the mycotoxin fumonisin B1 in food samples, cocaine and several metabolites in human urine, as well as several pesticides, antibiotics and drugs of abuse in urban wastewater.
Introduction
Nowadays, liquid chromatography (LC) hyphenated to mass spectrometry (MS) using a variety of mass analyzers is the technique of choice for the investigation of organic contaminants in most analyte/sample matrix combinations in environmental, food or toxicology fields. Mass analyzers used include triple quadrupole (QqQ),1–6 time-of-flight (TOF), hybrid quadrupole time-of-flight (QTOF),2,7–11 quadrupole-linear ion trap (QLIT)3,12,13 or Orbitrap.14 Many examples can be found in the literature dealing with pesticide residue analysis in environmental,15food16,17 or biological samples,18 using LC-MS based methods. Emerging contaminants, such as pharmaceuticals1,5 or drugs of abuse,19 amongst others, are increasingly being monitored in the environment by LC-MS because their medium-to-high polarity and low volatility make their determination fit better with LC. Similarly this applies to metabolites and transformation products, which are generally more polar than their parent molecules.LC-tandem MS (LC-MS/MS) operating in Selected Reaction Monitoring mode (SRM) with QqQ analysers are the workhorses nowadays in target analysis.20LC-MS/MS methods rarely include more than two hundred analytes,2,21,22 and with a few exceptions,23 most of them are focused on a single family of contaminants. Excellent sensitivity and notable selectivity are achieved by LC tandem MS, allowing reliable quantification and identification of a considerable number of compounds. However, the presence of other contaminants that might be present in the samples would be ignored in LC-MS/MS under SRM mode (the most common approach), due to the analyte-specific information acquired. There is a need in the field of public health to develop reliable methods for large-scale screening that are capable of detecting and identifying a large number of hazardous compounds that can potentially be present in environmental and food samples. For this purpose, full spectrum acquisition techniques capable of providing accurate mass measurements are a great help.
To solve the limitations of unit resolution mass spectrometers, two main alternatives, based on the use of high-resolution MS instruments, are of note at present: the time-of-flight24 and Orbitrap25,26 analysers. Both provide full spectrum accurate-mass data at satisfactory sensitivity. These capabilities are very helpful for detecting and identifying not only priority known pollutants but many other unknown contaminants that might be a risk for human health.27–29
Although quantitative applications have been reported using LC-TOF MS or LC-QTOF MS,8,10,30 quantification does not seem to be the most attractive feature of these analysers. This may be due to the higher limits of detection and narrower linear dynamic range in comparison to QqQ analysers. One of the most interesting applications of TOF MS deals with the wide-scope screening of a large number of contaminants and residues in different types of samples, as that allows a significant amount of useful information on ionisable compounds present in the sample to be obtained.31 Generic (universal) sample treatments and chromatographic separations are required to broaden the scope of the method to as many compounds as possible. Besides, the elevated acquisition speed of TOF makes it compatible with ultra-high pressure (Ultra-Performance) liquid chromatography (UHPLC/UPLC). This technique provides fast, high-resolution separations that will hopefully minimize matrix effects and render high mass spectra purity, improving the screening process.
Different strategies can be used to extract analytical information from full-acquisition accurate mass data. A genuine non-target analysis involves the automated component detection from the total ion chromatogram (TIC) and the mass spectra deconvolution for a subsequent comparison with mass spectral libraries. Nevertheless, electrospray ionization (ESI) is not an ion source as stable and reproducible as electron ionization,32 and commercial, standardized ESI mass spectra library are not available. Instead, theoretical mass spectra libraries, based on the molecular formula database, can be built which facilitate increasing the number of compounds that can be searched. These use accurate mass measurements and isotopic pattern information for identification. Home-made empirical libraries can also be used, but these normally include much fewer compounds due to the need to inject standards. These experimental libraries offer fragmentation and retention time information as well, providing more confidence in the compound identification process.33 However, the possibility of detecting and identifying the sample contaminants, using both mass spectra libraries in a non-target analysis, depends on the success of the deconvolution process, i.e. the capability of the software to find the component peaks and to obtain mass spectra as free as possible of sample interferents. Obviously, the more complex the matrix, the more difficult the deconvolution will be.
An efficient approach to overcome the component detection limitations is the use of “post-target” methodology,34,35i.e. the selection of the analytes to be searched is done after MS acquisition. A post-target screening facilitates the detection of the compounds as it is only focussed on those pollutants selected. It is unnecessary to totally deconvolute all components present in the samples, these mainly belong to matrix compounds. Furthermore, processing and reviewing steps become easier as fewer compounds are searched for and consequently detected.
In this work three sample types have been selected (wastewater, food and human urine) to explore the potential of UHPLC-(Q)TOF MS to detect and identify/elucidate organic contaminants and/or residues. Two strategies have been applied for this purpose: a post-target screening, based on mass filtering at the exact mass of the compound investigated (typically the (de)protonated molecule) using narrow mass extraction windows and a non-target methodology using both empirical and theoretical mass spectra libraries. QTOF MS has been used under MSE mode, i.e. simultaneous acquisition at low (LE) and high collision energy (HE) functions, which provides useful information on the (de)protonated molecules (commonly at LE) and on the main fragments ions (commonly in HE). On the basis of this information, and on isotopic distribution observed in the spectra, the reliable identification of the compounds detected in the samples was feasible.
Experimental
Reagents and chemicals
HPLC-grade water was obtained from deionized water passed through a Milli-Q water purification system (Millipore, Bedford, MA, USA). HPLC-grade methanol (MeOH) and acetonitrile (ACN) were purchased from ScharLab (Barcelona, Spain). Formic acid (HCOOH) (>98%) was obtained from Fluka (Buchs, Switzerland). Sodium hydroxide (>99%) was obtained from ScharLab. Leucine enkephalin, used as lock mass, was purchased from Sigma Aldrich (St Louis, MO, USA).Reference compounds were purchased from Acros Organics (Geel, Belgium), Bayer Hispania (Barcelona, Spain), Fort Dodge Veterinaria (Gerona, Spain), Vetoquinol Industrial (Madrid, Spain), Aventis Pharma (Madrid, Spain), Sigma Aldrich (St Louis, MO, USA), Cerilliant (Round Rock, TX, USA), Dr Ehrenstorfer (Augsburg, Germany), Riedel-de Haën (Seelze, Germany), the National Measurement Institute (Pymble, Australia) and Fluka. All reference materials presented purity higher than 93% (w/w).
Instrumentation
An UPLC Acquity system coupled with a hybrid quadrupole orthogonal acceleration-time-of-flight (Q-oaTOF) mass spectrometer (QTOF Premier, Waters, Milford, MA) provided with an orthogonal Z-spray lockspray electrospray interface (ESI) was used.Mobile phases A and B were water and methanol respectively, both with 0.01% formic acid. The separation was performed on an Acquity C18 BEH analytical column (150 mm × 2.1 mm, i.d. 1.7 μm) at a flow rate of 0.3 mL min−1 (at 60 °C). The initial percentage of methanol was 10%, which was linearly increased to 90% in 14 min, followed by a 2 min isocratic period and, then, returned to initial conditions during 2 min in total run duration of 18 min. The injection volume was 50 μL.
Cone and nebulizer gas were nitrogen (Praxair, Valencia, Spain) at flow rates of 60 L h−1 and 600 L h−1, respectively. The nitrogen desolvation temperature was set to 350 °C and the source temperature to 120 °C. A cone voltage of 25 V and capillary voltages of 3.5 kV and 2.5 kV in positive and negative ionisation modes, respectively, were used.
TOF MS resolution was ∼10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 at full width half maximum (FWHM) in V-mode. MS spectra were acquired over an m/z range 50–1000. Collision gas was argon 99.995% (Praxair, Valencia, Spain), which was always turned on with a pressure of approximately 5 × 10−3 mbar.
000 at full width half maximum (FWHM) in V-mode. MS spectra were acquired over an m/z range 50–1000. Collision gas was argon 99.995% (Praxair, Valencia, Spain), which was always turned on with a pressure of approximately 5 × 10−3 mbar.
Two acquisition functions were created with different collision energies. The first one, the low energy (LE) function, at low collision energy (4 eV) and the second one, the high energy (HE) function, with a collision energy ramp ranging from 15 to 40 eV. The scan time values of LE and HE functions were set to 0.2 and 0.15 s, respectively, both with an inter-scan delay of 0.05 s.
The lock mass (leucine enkephalin, 2 mg L−1 in ACN:water, 50:50) was introduced via the lock spray needle at a flow rate of 30 μL min−1 using a reagent manager pump (Waters). A cone voltage of 60–70 V was selected and checked daily to obtain adequate signal intensity for this compound (around 500 counts).
Calibration of the m/z-axis was performed using the built-in single-syringe pump, directly connected to the interface. Calibration from 50 to 1000 m/z was conducted with a 1:1 mixture of 0.05 M NaOH:5% HCOOH diluted (1:25) with water/ACN (20:80 v/v) plus imazalil (m/z 297.0561) at a final concentration of 500 μg L−1.
Data station operating software was MassLynx v 4.1. ChromaLynx XS application manager was used for non-target (deconvolution and library search) as well as for target analysis.
Sample treatment
8 wastewater samples—4 influent (IWW) and 4 effluent (EWW)—10 human urine and 6 food samples (2 oranges, 2 banana and 2 corn samples) were analysed for comparing the screening approaches.50 mL of wastewater were pre-concentrated by off-line SPE using 200 mg Oasis HLB cartridges, eluted with 5 mL of MeOH, evaporated under a gentle nitrogen stream at 40 °C and reconstructed with 1 mL water:MeOH (90:10 v/v).
Food sample extraction was performed according to previous work developed by our group.6,36 20 g of triturated and homogenized orange or banana samples were extracted with 60 mL water:MeOH (20:80 v/v) for 2 min using a high-speed blender, filtered and diluted with water:MeOH (20:80 v/v) to a final volume of 100 mL. Afterwards, an aliquot of the extract was diluted eightfold with water.
2.5 g of crushed corn sample were extracted with 10 mL ACN:water (80:20 v/v) with 0.1% HCOOH and mechanically shaken for 90 min.6 Afterwards, the solution was centrifuged, and a 5 mL aliquot of supernatant was diluted twofold with water.
Human urine samples from healthy volunteers and from people involved in drug detoxification programmes were centrifuged, diluted fivefold with water and directly injected into the LC-QTOF instrument.
Software parameters
The deconvolution and spectra rejection parameters were selected as follows:• minimum peak width at 5% height: 4 s,
• peak-to-peak baseline noise: 5,
• smoothing activated,
• mass tolerance (mass window width): 20 mDa,
• two mass chromatograms extracted for each component in LE function (5 mass chromatograms for HE), i.e. 2 or 5 coeluting ions to be extracted with the narrow window mass selected (±10 mDa).
The values for minimum peak width and mass window were selected as a function of the chromatographic resolution and mass accuracy data of our instrument.
Accurate mass scoring parameters were selected as follows:
• Number of ions used for accurate mass scoring: 2
• Minimum intensity (% of largest peak in the range): 10
• High precision mass tolerance (colouring in green): 2.5 mDa
• Low precision mass tolerance (colouring in yellow) = 5 mDa
These values were selected according to our own experience and characteristics of the LC-QTOF MS equipment used, but they might be modified according to the performance of the instrument used in each laboratory.
Results and discussion
Mass resolving power is an important issue for the correct detection and identification of the suspect compounds. Even if the 10000 at 10% valley resolution (20000 FWHM) required by the EC Decision 2002 (ref. 37) is not achieved by the (Q)TOF mass spectrometer used, we consider that mass accuracy is really the key in the identification of the compounds in the wide-scope screening. Although strongly correlated, mass resolving power and mass accuracy are not strictly the same. In a previous work,33 improving the resolution (about 18000 FWHM) by doubling the path length using the so-called W-mode in different matrices (influent and effluent wastewater, surface water, pepper and cucumber) showed no significant effect for the compounds tested on mass accuracy achieved using UHPLC separation. On the other hand, a 20 mDa mass window has been used in this work for both, non-target and post-target strategies, as a compromise between ensuring correct chromatographic peak at both ends and attainable selectivity. Lower mass windows (e.g. 5 and 10 mDa) were also tested, but finally discarded as no satisfactory chromatographic peaks were always ensured. Newer instruments with stable mass accuracy across the peak could facilitate the screening process by reducing this mass window, even down to 1 mDa.
The non-target and post-target strategies studied in this work were applied to all selected samples to test the screening capabilities and for comparison purposes. A flowchart of the process is shown in Fig. 1.
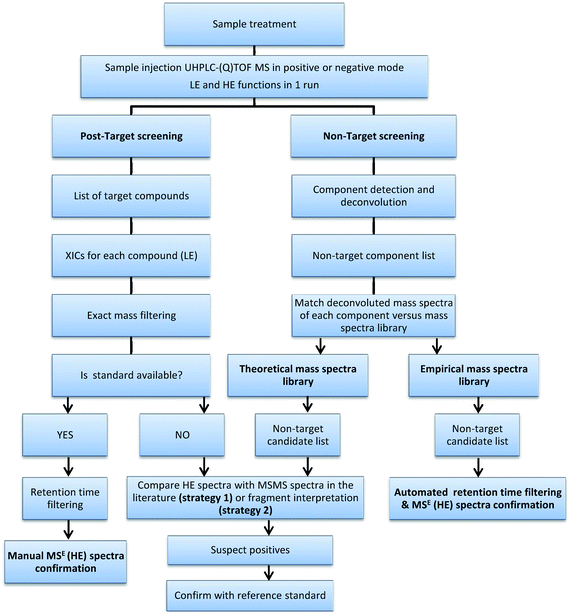 | ||
| Fig. 1 Flowchart of the overall screening process. | ||
1. Non-target screening
A true non-target screening using LC-(Q)TOF MS is a challenging task as it is very difficult to detect and identify trace level contaminants when no selection is made on the compounds to be searched.38 In this work, non-target screening was applied to environmental, food and biological samples to evaluate the potential of the algorithm to detect components when dealing with complex matrices. For this purpose, the deconvolution software ChromaLynx XS in a non-target mode was used. The software applies a component detection algorithm (CODA) to deconvolute the TIC and detect the components present in the sample. Afterwards, it compares the spectra assigned to every component with those included in the home-made libraries. To facilitate the confirmation of the identity of the components detected, two functions were simultaneously acquired at different collision energies (MSE). The LE function was used to obtain the (de)protonated molecules (occasionally adducts and fragment ions). The HE function was used to promote fragmentation, improving the identification of the positive findings as spectra obtained were quite similar to those of MS/MS experiments.33,39 This acquisition provides reproducible spectra without the need of precursor ion pre-selection in the first quadrupole. The success for detecting and identifying non-target compounds using this approach obviously depends on the deconvolution process. In addition, MSE provides not only fragmentation spectra but also isotopic pattern information of the fragments and it conserves adduct and/or dimer information. However, two main limitations were noticed when MSE was applied to non-target screening:(a) As there is no pre-selection of precursor ion in the quadrupole, the MSE approach is less specific and might be conflictive when dealing with non-selective fragments in the presence of co-eluting related compounds. This occurs, for example, when investigating amphetamine-like compounds amphetamine and methamphetamine. As can be seen in Fig. 2, both drugs elute at very close retention times and present poor and identical HE spectra, with the most abundant ion being the non-selective fragment at m/z 91 corresponding to tropylium ion. Moreover, as protonated molecules have relatively poor abundance in the LE function (especially amphetamine) it could be very difficult to distinguish both compounds at low concentration levels.
![LE and HE mass spectra for amphetamine-like compounds. LE spectra for methamphetamine and amphetamine show notable in-source fragmentation with different [M + H]+ ion (m/z 150.1290 and 136.1134). Identical HE spectra (m/z 91.0548 corresponding to tropylium ion) are obtained for both compounds.](/image/article/2012/AY/c1ay05385j/c1ay05385j-f2.gif) | ||
| Fig. 2 LE and HE mass spectra for amphetamine-like compounds. LE spectra for methamphetamine and amphetamine show notable in-source fragmentation with different [M + H]+ ion (m/z 150.1290 and 136.1134). Identical HE spectra (m/z 91.0548 corresponding to tropylium ion) are obtained for both compounds. | ||
(b) The success of the MSE approach can be limited by the quality of the spectrum.39 Thus, low sensitivity or strongly interfered spectra end up making it unfeasible to match with library spectra as well as not being able to elucidate the component using fragment interpretation. In these cases, additional MS/MS experiments would be helpful in the identification/elucidation process.
Finally, the software returns a match factor for the comparison of standard and candidate mass spectra and gives the mass errors for the 2 most abundant ions present in the LE function and for the 5 main fragment ions present in the HE function. A positive match can be filtered by a minimum match factor and retention time, if available. In this work, a relatively low match factor (in reverse fit) of 70% was selected as a compromise. This facilitated the reviewing process of positives without losing potential hazardous compounds that could be present in the samples although with low match factors. Two types of mass spectra libraries were evaluated in this work as discussed in the following sections.
When no or unsatisfactory match is obtained, the components appear to be tentative. In these cases, the elucidation of the compound requires a lot of time and effort with a low possibility of success. Furthermore, the majority of non-matched components are likely to be matrix compounds.
A drawback of the theoretical library (and also of the empirical mass spectra library) is that TOF MS spectra are stored in nominal mass for NIST format compatibility, and in this step the mass accuracy information given by TOF MS is lost. In order to minimize this limitation, the mass errors between the measured masses of the compound detected and the exact masses of the candidates formulae are calculated and used in a subsequent step, to rank them and to propose the most plausible identity (accurate mass scoring).35
Detection/identification problems derived from LE adducts formation and/or important in-source fragmentation were prevented by analyzing the samples under exactly the same conditions as the reference standards. This favoured the task and minimized the risk of potential false negatives. Furthermore, HE mass spectra were automatically matched with those included in the empirical library which greatly facilitated the confirmation of the compound identity. In our experience, HE provided highly reproducible spectra (independently of the type of sample analysed) when the component was found at relatively high abundance. As signal intensity is the main limitation during the component detection step, HE spectra facilitated identification of the compound when its spectrum was available in the library in those components detected by the non-target approach.
2. Post-target screening
Trying to avoid the dependence of the screening success on the component detection algorithm, a post-target screening strategy was applied including an extraordinarily large number of compounds in the search. The term “post-target” was first used by our group34,35,40 as a target screening without pre-selection of the analytes before analysis. It consists of searching for a list of target compounds after MS full-acquisition. Other authors name this approach, when reference standards are unavailable, as suspect screening.20 In the post-target screening, a database with the same compounds included in the theoretical library of the non-target approach was used (ESI†). ChromaLynx XS uses the molecular formula to calculate the exact mass for [M + H]+. Then, the software automatically performs the extraction of a nw-XIC (20 mDa) for each compound in the LE and HE functions and looks for peaks (S/N and peak width higher than pre-selected values) in the corresponding chromatogram. A list of potential candidates found in the sample is shown in different colours depending on accurate mass measurement; positive (green) for error <2.5 mDa, tentative (yellow) for error between 2.5 and 10 mDa, and negative (red) for error >10 mDa. Furthermore, as in the non-target approach, ChromLynx XS filters positive findings according to retention time deviation limit when this information is available in the database (reference standards previously injected). The retention time window was set in ±0.5 min but accepted tolerance was 2.5%. Thus, the retention times for 231 analytes, injected when building the empirical library, were also introduced in the database. In this way, nw-XIC, top peak spectra and mass error as well as isotopic distribution fit (i-FIT) information, retention time (measured and expected when already known) and peak area were available for positive matches. Fig. 3 shows the ChromaLynx XS browser for a positive of mycotoxin Fumonisin B1 in a corn sample using this approach.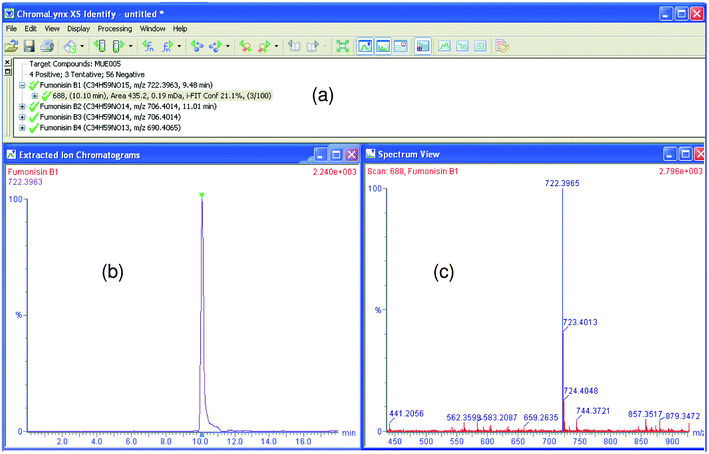 | ||
| Fig. 3 ChromaLynx XS browser with accurate mass confirmation for Fumonisin B1 in corn using post-target screening. (a) Candidate list for compound with mass error <2.5 mDa (which offers retention time, area, mass error and i-FIT information), (b) nw-XIC for suspected candidate (at 20 mDa window). (c) mass spectrum (in blue, candidate peak is shown). | ||
QTOF MS post-target screening has proved to be an efficient tool due to the high number of pollutants screened. The potential of this approach to detect different families of organic contaminants, for example drugs of abuse or antibiotics in environmental samples, has been reported recently.39,41 The easy reviewing step and the relevant information obtained, such as accurate mass spectrum of the peak, mass error for the protonated molecule and the most abundant fragments, and isotopic distribution, give high confidence to the confirmation of potential positives even without reference standards being available. The large number of contaminants included in the list (more than 1000) opens a new scenario in screening, favouring a more realistic overview when investigating organic contaminants in different applied fields. However, if only the predicted presence of the protonated molecule was taken into account in the LE function, potential in-source fragments would not be detected (e.g. as occurs in amphetamine-like compounds, see Fig. 2), not even sodium or other adducts that could be formed.
In this work, formic acid was added to the mobile phases. Under this situation, ammonia adducts and other adducts like [M + MeOH + H]+ or [M + K]+ would not normally be expected. However, sodium adducts are common for many LC-amenable compounds, and they might be present despite using formic acid. In our own experience, 88 out of 231 compounds (38%) included in the experimental library showed sodium adducts at relative abundance higher than 10%. Among them, 38 compounds (16% of the total compounds) presented the [M + H]+ ion at relative intensity lower than 10%, this becoming the [M + Na]+, the most abundant ion in the mass spectra. When analyzing real samples, sodium adducts might be found at higher abundance due to the normal presence of sodium in the sample matrices. Therefore, it is important to include sodium adducts in the screening to avoid potential false negatives in those cases where it is the most abundant ion (see Table 1). However, it seems reasonable not to include sodium adducts for all analytes investigated, as the processing and the reviewing step would be much longer and more tedious. The injection of reference standards and/or literature search, along with analyst experience, are necessary parts of knowing when it is reasonable to include compound adducts to improve the confidence of the screening process.
| Effluent wastewater | Target screening | Non-target screening | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Tentative identification | Confirmed with standard? | LE function | Theoretical library | Empirical library | ||||||
| Compound | RT (min) | ΔRT (%) | Δmass/mDa | Strategy 1a | Strategy 2b | Na adduct? | Fragment ions? | Match reverse fit | Match reverse fit | |
| a Strategy 1, used for tentative identification of the compounds when the standard was not available, consisted of comparing the main fragments observed in the HE function with common MS/MS product ions reported in the literature. b Strategy 2 was made by justifying the HE accurate mass fragments using a bond-disconnecting software. | ||||||||||
| Antipyrine | 5.43 | — | 1.4 | — | ||||||
| Bamethan | 3.43 | — | 0.5 | ✓ | × | — | ||||
| Bisoprolol | 7.25 | — | 0.0 | ✓ | ✓ | — | 929 | |||
| Caffeine | 4.19 | — | 0.2 | ✓ | ✓ | — | ||||
| Carbendazim | 4.45 | 0.04 | 0.5 | Yes | ✓(1) | 923 | 898 | |||
| Celiprolol | 6.41 | — | 0.7 | ✓ | ✓ | — | ||||
| Clarithromycin | 10.11 | 0.10 | 1.7 | Yes | 855 | |||||
| Clofibric acid | 12.60 | — | 2.5 | ✓ | × | — | ||||
| Codeine | 2.82 | 0.00 | 1.6 | Yes | 943 | 794 | ||||
| Diazinon | 13.04 | 0.01 | 0.9 | Yes | ||||||
| Diuron | 9.98 | 0.02 | 0.3 | Yes | ||||||
| Erithromycin (−H2O) | 9.86 | 0.01 | 1.2 | Yes | ||||||
| Gabapentin | 3.47 | 0.09 | 0.5 | ✓ | ✓ | Yes | ✓(2) | 903 | ||
| Irbesartan | 11.42 | 0.10 | 0.0 | ✓ | ✓ | Yes | 953 | |||
| Ketoprofen | 10.61 | 0.04 | 0.5 | Yes | ✓ | |||||
| Metoprolol | 5.55 | — | 0.0 | ✓ | ✓ | — | ||||
| Nordiazepam | 11.01 | — | 0.1 | ✓ | — | |||||
| OD-PABA | 5.49 | — | 0.9 | ✓ | ✓ | — | 953 | |||
| Oxazepam | 10.19 | — | 0.4 | ✓ | — | |||||
| Oxprenolol | 2.77 | — | 0.2 | ✓ | ✓ | — | ||||
| Propylphenazone | 9.29 | — | 0.4 | ✓ | ✓ | — | ||||
| Terbutryn | 11.69 | 0.14 | 1.0 | Yes | ||||||
| Thiabendazol | 5.22 | 0.12 | 0.1 | Yes | ||||||
| Trimethoprim | 3.83 | 0.10 | 0.9 | Yes | 980 | 925 | ||||
| Valsartan | 11.59 | 0.05 | 1.9 | ✓ | ✓ | Yes | ✓ | 849 | ||
| Venlafaxine | 7.17 | 0.46 | 0.3 | Yes | ✓(1) | |||||
| MDMA | 3.78 | 0.00 | 0.0 | Yes | ✓(2) | |||||
| Bezafibrate* | 11.07 | 0.01 | 2.1 | Yes | ✓ | |||||
| Gemfibrozil* | 13.84 | 0.03 | 2.5 | Yes | ✓ | |||||
| *Compounds found only as sodium adduct ion | ||||||||||
| Orange | Target screening | Non-target screening | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Tentative identification | Confirmed with standard? | LE function | Theoretical library | Empirical library | ||||||
| Compound | RT (min) | ΔRT (%) | Δmass/mDa | Strategy 1a | Strategy 2b | Na adduct? | Fragment ions? | Match reverse fit | Match reverse fit | |
| Imazalil | 9.22 | 0.03 | 0.3 | Yes | 916 | 925 | ||||
| Thiabendazol | 5.21 | 0.03 | 0.3 | Yes | 893 | 916 | ||||
| Banana peel | Target screening | Non-target screening | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Tentative identification | Confirmed with standard? | LE function | Theoretical library | Empirical library | ||||||
| Compound | RT (min) | ΔRT (%) | Δmass/mDa | Strategy 1ab | Strategy 2b | Na adduct? | Fragment ions? | Match reverse fit | Match reverse fit | |
| Chlorpyrifos | 14.61 | 0.01 | 0.5 | Yes | ✓ | |||||
| Diazinon | 13.03 | 0.01 | 0.7 | Yes | 854 | 863 | ||||
| Imazalil | 9.21 | 0.02 | 0.9 | Yes | 931 | 923 | ||||
| Corn | Target screening | Non-target screening | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Tentative identification | Confirmed with standard? | LE function | Theoretical library | Empirical library | ||||||
| Compound | RT (min) | ΔRT (%) | Δmass/mDa | Strategy 1ab | Strategy 2b | Na adduct? | Fragment ions? | Match reverse fit | Match reverse fit | |
| Fumonisin B1 | 10.10 | 0.03 | 0.8 | Yes | 785 | 800 | ||||
| Fumonisin B2 | 11.69 | 0.03 | 1.2 | Yes | 681 | 686 | ||||
| Fumonisin B3 | 10.97 | — | 1.3 | ✓ | ✓ | — | ||||
| Fumonisin B4 | 12.44 | — | 0.2 | ✓ | ✓ | — | ||||
| Urine sample | Target screening | Non-target screening | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Tentative identification | Confirmed with standard? | LE function | Theoretical library | Empirical library | ||||||
| Compound | RT (min) | ΔRT (%) | Δmass/mDa | Strategy 1ab | Strategy 2b | Na adduct? | Fragment ions? | Match reverse fit | Match reverse fit | |
| Gabapentin | 3.27 | 0.05 | 0.8 | Yes | (2) | 893 | ||||
| Nicotine | 2.72 | — | 1.1 | ✓ | ✓ | — | ||||
| Paracetamol | 2.68 | 0.02 | 0.3 | Yes | ||||||
| Risperidone | 6.72 | 0.10 | 0.0 | Yes | ||||||
| Amphetamine | 3.27 | 0.20 | 0.4 | Yes | (2) | 874 | ||||
| Benzoylecgonine | 5.04 | 0.02 | 1.0 | Yes | 895 | 967 | ||||
| Cocaethylene | 6.25 | 0.11 | 0.4 | Yes | ||||||
| Cocaine | 5.16 | 0.19 | 0.8 | Yes | 716 | |||||
| NorBenzoylecgonine | 5.28 | 0.00 | 0.6 | Yes | ||||||
| NorCocaethylene | 5.71 | — | 0.7 | ✓ | ✓ | — | ||||
| NorCocaine | 5.34 | 0.05 | 0.2 | ✓ | ✓ | — | ||||
A similar problem may occur when important in-source fragmentation takes place at the LE function. In this work, we used 25 V cone voltage as better sensitivity was observed for selected analytes in the 20–30 V range.33 Obviously, this cone voltage is a compromise value as it is not the best choice for all compounds but it is impossible to optimize any variable for all LC-amenable compounds included in the database. As previously stated, amphetamine ([M + H]+ 136.1126) presents an in-source fragment at m/z 91.0553 as the most abundant ion in the spectra, while the protonated molecule has an abundance lower than 10% (Fig. 2). Other examples are the insecticide carbaryl (fragment at m/z 145.0563) or pesticide metabolite aldicarb sulfoxide (fragment at m/z 89.0415). In these cases, analyte detection in samples based on testing [M + H]+ presence would be only feasible at relatively high analyte concentrations.
Other compounds, like anabolic steroids, are frequently ionised forming adducts with MeOH, acetonitrile, ammonium or sodium (as a function of the mobile phase and sample matrix composition) and/or they suffer in-source fragmentation with neutral losses of one, or even two, water molecules ([M − H2O + H]+, [M − 2H2O + H]+).42 The later drawback is more difficult to solve than adducts formation, but it could be circumvented by including empirical formula of the known fragment ions in the database. Again, information reported in the literature and/or from reference standards injection would be required to include expected fragments in the database. Although fragmentation behaviour is not completely known in most cases, in our experience, this effect is less common than adduct formation. Indeed, only 6 out of 231 compounds (3% of the compounds studied) almost exclusively presented the fragment ion as base peak, with the protonated ion being practically absent. In these particular cases, monitoring this fragment is mandatory for compound detection. In-source fragmentation turns into a useful confirmatory tool when the reference standard is available and/or its behaviour is well known. Thus, including most abundant fragments is always useful for automated confirmation.
As an illustrative example, Table 2 shows information on database entries for different types of analytes included in this work. The molecular formula of the ion, when adduct formation and/or in-source fragmentation occurred, was also introduced in the database, as well as the bibliographic source, when information on possible occurrence of these ions was not directly obtained from reference standard injection.
| Compound | Molecular formula | Rt (min) | Ion type | Accurate mass | Pollutant family | Source |
|---|---|---|---|---|---|---|
| Oxytetracycline* | C22H24N2O9 | 4.83 | [M + H]+ | 461.1560 | Antibiotic | |
| Oxytetracycline F1 | C22H22N2O8 | 4.83 | Fragment ion | 443.1454 | Antibiotic | |
| Oxytetracycline F2 | C22H19NO8 | 4.83 | Fragment ion | 426.1189 | Antibiotic | |
| Amphetamine | C9H13N | 3.06 | [M + H]+ | 136.1126 | Illicit drug | |
| Amphetamine F1 | C9H10 | 3.06 | Fragment ion | 119.0861 | Illicit drug | |
| Amphetamine F2* | C7H6 | 3.06 | Fragment ion | 91.0548 | Illicit drug | |
| MDMA | C11H15NO2 | 3.14 | [M + H]+ | 194.1181 | Illicit drug | |
| MDMA F1* | C10H10O2 | 3.14 | Fragment ion | 163.0759 | Illicit drug | |
| MDMA F2 | C8H6O2 | 3.14 | Fragment ion | 135.0446 | Illicit drug | |
| 6-OH-4-Cl-dehydromethyltestosterone | C20H27O3Cl | 9.91 | [M + H]+ | 351.1727 | Steroid | |
| 6-OH-4-Cl-dehydromethyltestosterone (–H2O) | C20H25O2Cl | 9.91 | Fragment ion | 333.1621 | Steroid | |
| 6-OH-4-Cl-dehydromethyltestosterone (−2 × H2O) | C20H23OCl | 9.91 | Fragment ion | 315.1515 | Steroid | |
| 6-OH-4-Cl-dehydromethyltestosterone (Na)* | C20H26NaO3Cl | 9.91 | [M + Na]+ | 373.1547 | Steroid | |
| Ethisterone | C21H28O2 | — | [M + H]+ | 313.2168 | Steroid | JMS,42,2007,497-516 |
| Ethisterone [M + Na + MeOH]+ | C22H31NaO3 | — | [M + Na + MeOH]+ | 367.2249 | Steroid | JMS,42,2007,497-516 |
| Fumonisin B1* | C34H59NO15 | 10.13 | [M + H]+ | 722.3963 | Mycotoxin | |
| Fumonisin B2* | C34H59NO14 | 11.66 | [M + H]+ | 706.4014 | Mycotoxin | |
| Aldicarb sulfoxide | C7H14N2O3S | 3.19 | [M + H]+ | 207.0803 | Pesticide | |
| Aldicarb sulfoxide (Na)* | C7H13NaN2O3S | 3.19 | [M + Na]+ | 229.0623 | Pesticide | |
| Aldicarb sulfoxide F1* | C4H8S | 3.19 | Fragment ion | 89.0351 | Pesticide | |
| Aldicarb sulfoxide F2 | C5H9NOS | 3.19 | Fragment ion | 132.0483 | Pesticide | |
| Tebufenozide | C22H28N2O2 | 12.54 | [M + H]+ | 353.2229 | Pesticide | |
| Tebufenozide (Na)* | C22H27N2O2Na | 12.54 | [M + Na]+ | 375.2048 | Pesticide | |
| Tebufenozide (2M + Na)* | C44H55N4O4Na | 12.54 | [2M + Na]+ | 727.4199 | Pesticide | |
| Tebufenozide F1* | C18H20N2O2 | 12.54 | Fragment ion | 297.1603 | Pesticide | |
| Tebufenozide F2* | C9H8O | 12.54 | Fragment ion | 133.0653 | Pesticide | |
| Azinphos-methyl | C10H12N3O3PS2 | 10.49 | [M + H]+ | 318.0136 | Pesticide | |
| Azinphos-methyl (Na)* | C10H11N3O3PS2Na | 10.49 | [M + Na]+ | 339.9956 | Pesticide | |
| Azinphos-methyl F1 | C8H5N3O | 10.49 | Fragment ion | 160.0511 | Pesticide | |
| Azinphos-methyl F2* | C8H5NO | 10.49 | Fragment ion | 132.0449 | Pesticide | |
| Azoxystrobin | C22H17N3O5 | 10.97 | [M + H]+ | 404.1246 | Pesticide | |
| Azoxystrobin (Na)* | C22H16NaN3O5 | 10.97 | [M + Na]+ | 426.1066 | Pesticide | |
| Azoxystrobin F1* | C21H13N3O4 | 10.97 | Fragment ion | 372.0984 | Pesticide | |
| Bifenazate | C17H20N2O3 | 11.92 | [M + H]+ | 1.0078 | Pesticide | |
| Bifenazate (Na)* | C17H19N2O3Na | 11.92 | [M + Na]+ | 22.9898 | Pesticide | |
| Bifenazate F1 | C13H11NO | 11.92 | Fragment ion | 198.0919 | Pesticide | |
| Bifenazate F2 | C12H11N | 11.92 | Fragment ion | 170.0970 | Pesticide | |
| Dimethoate | C5H12NO3PS2 | 5.76 | [M + H]+ | 230.0075 | Pesticide | |
| Dimethoate (Na)* | C5H11NaNO3PS2 | 5.76 | [M + Na]+ | 251.9895 | Pesticide | |
| Methiocarb sulfone | C11H15NO4S | 6.27 | [M + H]+ | 258.0800 | Pesticide | |
| Methiocarb sulfone (Na)* | C11H14NO4SNa | 6.27 | [M + Na]+ | 280.0620 | Pesticide | |
| Methiocarb sulfone F1 | C9H12O3S | 6.27 | Fragment ion | 201.0585 | Pesticide | |
| Methiocarb sulfone F2* | C8H9O | 6.27 | Fragment ion | 122.0732 | Pesticide | |
| Methiocarb sulfoxide | C11H15NO3S | 5.73 | [M + H]+ | 242.0851 | Pesticide | |
| Methiocarb sulfoxide (Na) | C11H14NO3SNa | 5.73 | [M + Na]+ | 264.0671 | Pesticide | |
| Methiocarb sulfoxide F1* | C9H12O2S | 5.73 | Fragment ion | 185.0636 | Pesticide | |
| Thiamethoxam | C8H10ClN5O3S | 4.26 | [M + H]+ | 292.0271 | Pesticide | |
| Thiamethoxam (Na)* | C8H9ClN5O3SNa | 4.26 | [M + Na]+ | 314.0091 | Pesticide | |
| Thiamethoxam F1* | C8H10N4OS | 4.26 | Fragment ion | 211.0654 | Pesticide | |
| Thiamethoxam F2 | C4H2NSCl | 4.26 | Fragment ion | 131.9675 | Pesticide | |
| Thiobencarb | C12H16ClNOS | 13.39 | [M + H]+ | 258.0719 | Pesticide | |
| Thiobencarb (Na)* | C12H15NaClNOS | 13.39 | [M + Na]+ | 280.0539 | Pesticide | |
| Thiobencarb F1* | C7H5Cl | 13.39 | Fragment ion | 125.0158 | Pesticide | |
| Thiodicarb | C10H18N4O4S3 | 9.36 | [M + H]+ | 355.0568 | Pesticide | |
| Thiodicarb (Na)* | C10H17NaN4O4S3 | 9.36 | [M + Na]+ | 377.0388 | Pesticide | |
| Thiodicarb F1 | C3H5NS | 9.36 | Fragment ion | 88.0221 | Pesticide | |
| Bezafibrate | C19H20ClNO4 | 11.06 | [M + H]+ | 362.1159 | Pharmaceutical | |
| Bezafibrate (Na)* | C19H19NaClNO4 | 11.06 | [M + Na]+ | 384.0979 | Pharmaceutical | |
| Chloramphenicol | C11H12Cl2N2O5 | 6.46 | [M + H]+ | 323.0201 | Pharmaceutical | |
| Chloramphenicol (Na) | C11H11NaCl2N2O5 | 6.46 | [M + Na]+ | 345.0021 | Pharmaceutical | |
| Chloramphenicol F1* | C11H10N2O4Cl2 | 6.46 | Fragment ion | 305.0101 | Pharmaceutical | |
| Chloramphenicol F2* | C10H8N2O3Cl2 | 6.46 | Fragment ion | 275.0002 | Pharmaceutical | |
| Chloramphenicol F3 | C11H8NOCl2 | 6.46 | Fragment ion | 241.0078 | Pharmaceutical | |
| Gemfibrozil | C15H22O3 | 13.81 | [M + H]+ | 251.1647 | Pharmaceutical | |
| Gemfibrozil (Na)* | C15H21NaO3 | 13.81 | [M + Na]+ | 273.1467 | Pharmaceutical |
3. Application to samples
After application of the screening strategies to selected food, wastewater and human urine samples, the post-target approach was found to be the most efficient for wide-scope screening. In all samples analyzed, the number of positives was higher than using the non-target approach, in this way giving a more realistic overview of the presence of organic pollutants in the samples. A summary of the results obtained for selected samples is shown in Table 1. The list of pollutants found by target and non-target screening (those with an adequate match reverse fit) is reported together with the main information managed (mass error and retention time), as well as retention time deviation when reference standard was available. Almost in all cases, Rt deviation was lower than 1%. However, the retention time window for positive match was ±0.5 min due to the wide range of matrix analysed having, in some particular cases, deviations higher than 2% typically accepted as in the case of Venlafaxin. Confirmation using MSE is also shown when it could be made. When reference standards were unavailable, information on fragmentation and retention time was absent. Two strategies were followed to improve the confidence in the compound identification.The first strategy (Strategy 1 in Table 1) was to simply compare main fragments observed in HE acquisition with common MS/MS product ions reported in the literature for the suspect compound. This was the case for the antibiotic gabapentin, which was detected and identified in urine and wastewater by the presence of two abundant fragments in the HE spectrum with m/z 137.0966 and 154.1232 (Fig. 4). These fragment ions were also present in the LE function and had been reported by other authors for determination of gabapentin by QqQ.43–45 Elemental composition for these two fragments was calculated based on their accurate masses obtaining errors of 0.7 and 0.2 mDa, respectively.
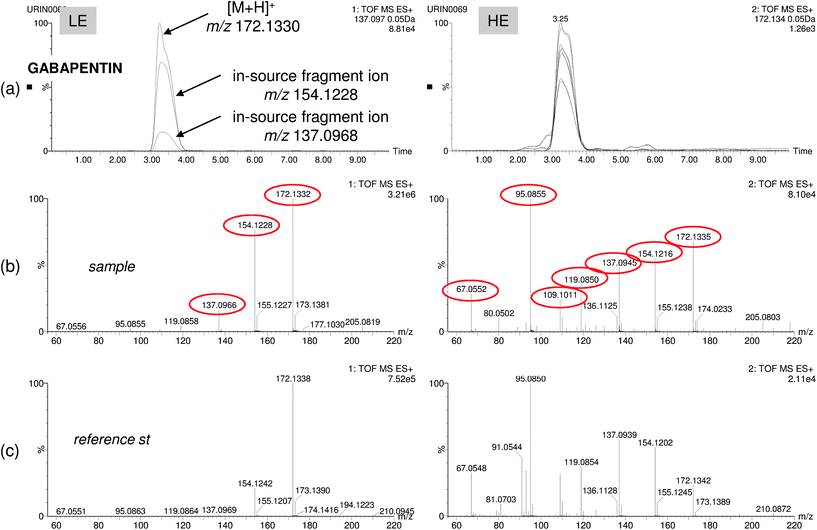 | ||
| Fig. 4 Positive finding of the pharmaceutical gabapentin in human urine: (a) overlapped nw-XIC for three main ions (protonated ion at m/z 172 and in-source fragments at m/z 154 and 137) in the LE function and seven coeluting ions in the HE function. LE and HE spectra for sample (b) and reference standard (c) showing good correlation for up to six abundant fragment ions. | ||
The second strategy (Strategy 2 in Table 1) consisted of justifying the fragments accurate mass (typically observed in the HE spectra) using MassFragment software. This software applies a bond-disconnecting methodology to obtain possible structures for the fragment ions from a given molecule. An example of this approach is shown in Fig. 5, where identification of main fragments of the pharmaceutical irbesartan was carried out. For this purpose, LE and HE combined spectrum of suspect irbesartan was extracted from the chromatographic peak (Fig. 5a and b). The main fragments were justified with the MassFragment tool obtaining reliable structures for all of them. In order to avoid spectrum interferences that could complicate the identification process, recognizing which ions are fragments and which are not, becomes mandatory. From this point of view, UHPLC resolution proved to be valuable for choosing perfectly coeluting ions (see Fig. 5c). Irbesartan is an angiostensin II receptor antagonist used in the treatment of hypertension that has been in the market for over 10 years.46 Some fragments observed for irbesartan had been previously reported by ion trap;46 the most used SRM transition coincides with the most abundant fragment ion of the TOF spectra (m/z 207.0922).47,48 However, as MassFragment is a bond-disconnecting software, correct justification is not always feasible. Thus, for m/z 192 ion unreliable structures were suggested. In these cases, previous analyst knowledge or better fragmentation prediction software is necessary.
![Positive finding of the pharmaceutical irbesartan in effluent wastewater. Spectra for LE function (a) and HE function (b) of the suspect peak and justification of the HE fragments using MassFragment software. (c) nw-XICs (20 mDa mass window) for [M + H]+ in LE function and main fragments in HE function.](/image/article/2012/AY/c1ay05385j/c1ay05385j-f5.gif) | ||
| Fig. 5 Positive finding of the pharmaceutical irbesartan in effluent wastewater. Spectra for LE function (a) and HE function (b) of the suspect peak and justification of the HE fragments using MassFragment software. (c) nw-XICs (20 mDa mass window) for [M + H]+ in LE function and main fragments in HE function. | ||
In Table 1, the strategy used for the identification of each suspected positive is shown. Bibliographic search and fragment interpretation were helpful to confirm potential positives. When a disagreement occurred between experimental and literature data for fragment ions (if available), and when structures provided by MassFragment software did not fit with the structure of the candidate, the suspected positive could not be confirmed, and no further research was performed for its elucidation.
Following the above mentioned strategies, high confidence in the identification process can be achieved. However, no definitive confirmation should be made without injecting the reference standard. Thus, for the most frequently detected pharmaceuticals, irbesartan, valsartan and gabapentin, the reference compounds were acquired. After injecting the standard solutions, all suspect positives in wastewater were confirmed. Our experience on identification of suspect organic contaminants by LC-QTOF under MSE mode is that the great majority of suspect positives (around 95%) were subsequently confirmed when the reference standard was acquired. This means that acquisition of expensive standards could be made only when solid evidence exists on their presence in samples analyzed. The decision on which standards should be acquired would then be made on the basis of previous findings by QTOF MS.
To overcome some post-target limitations and to enhance detectability and identification reliability, improvements in the database approach were made to minimize “missing” compounds due to abundant adduct formation and/or in-source fragmentation. More entries were added in the pollutant database for compounds with a high degree of fragmentation and sodium adduct formation. This is easier when information for the compound is available. After reprocessing the samples using the new, enlarged database, two more compounds (gemfibrozil and bezafibrate) were found in wastewater. These compounds were not detected before due to the abundant sodium adduct formation in positive electrospray ionisation (marked as * in Table 1). In addition, not only the detection step was improved but also the confidence in the identification, as for several analytes, both the protonated molecule and in-source fragments/sodium adducts were also detected (information shown in Table 1). To exemplify this feature, Fig. 6 shows a positive finding of MDMA in EWW. As can be seen, the protonated ion and main in-source fragment ion of the compound were both detected, the latter being much more abundant than the protonated molecule.
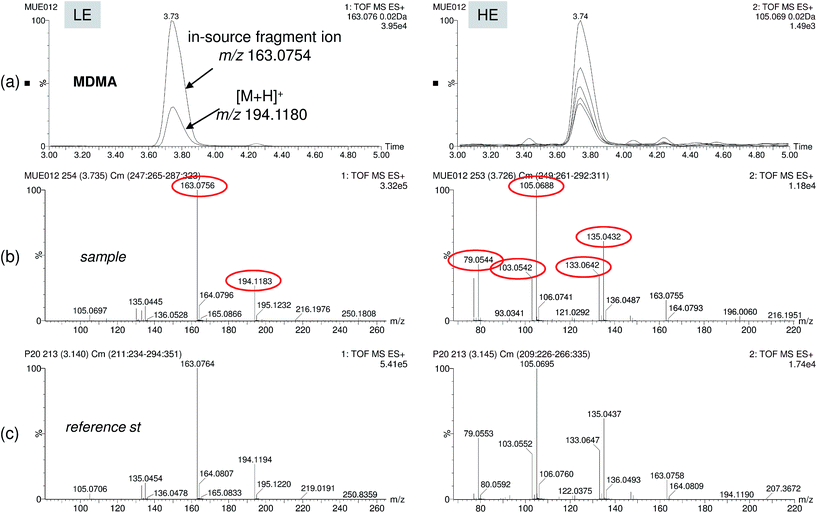 | ||
| Fig. 6 Positive finding of the drug of abuse MDMA in effluent wastewater. (a) Overlapped nw-XIC for two main ions (protonated ion at m/z 194 and in-source fragment at m/z 163) in the LE function, and up to five coeluting ions in the HE function. LE and HE spectra for sample (b) and reference standard (c) showing good correlation for up to five abundant fragment ions. | ||
As a summary, two situations could be considered when using the post-target approach based on QTOF measurements:
(a) Detection of target analytes for which standard is available and has been previously injected under the same conditions as the samples. In this case, retention time, in-source fragmentation and adduct formation became useful tools, making the confirmation of findings highly reliable, surely unequivocal.
(b) Detection of suspect compounds for which reference standards are unavailable. Obviously, the situation requires extra-work and time. After a careful study of the full-scan accurate mass data obtained for the suspect compound, a reliable identification could be advanced. A definitive confirmation by injection of the reference standard would be required in the case that significant environmental or legal implications were associated to the presence of the suspect compound. Here, the experience of the analyst and their background on mass spectrometry is of the utmost relevance.
Regarding the non-target screening results, it must be noted that the deconvolution process depends to a great extent on the intensity of the chromatographic peak. Using this approach, several contaminants were missed, as the number of compounds found in the samples was considerably lower than using the targeted one (Table 1). Furthermore, non-target screening with empirical library allowed us to detect very few compounds, not only because of the component detection limitations but also due to fewer entries in this library (231). However, confirmation of the identity becomes simultaneous and more reliable than with other approaches (i.e. theoretical library) as LE and HE spectra are compared with those included in the empirical library making unlikely the reporting of false positives. As an example, Fig. 7 shows a corn sample positive to fumonisin. In this figure, two and five coeluting ions were selected for component detection in the LE and HE functions, respectively (Fig. 7a). Both deconvoluted LE and HE mass spectra were automatically compared with those of fumonisin B1 included in the empirical mass spectra library with a match of 80% (Fig. 7b and c).
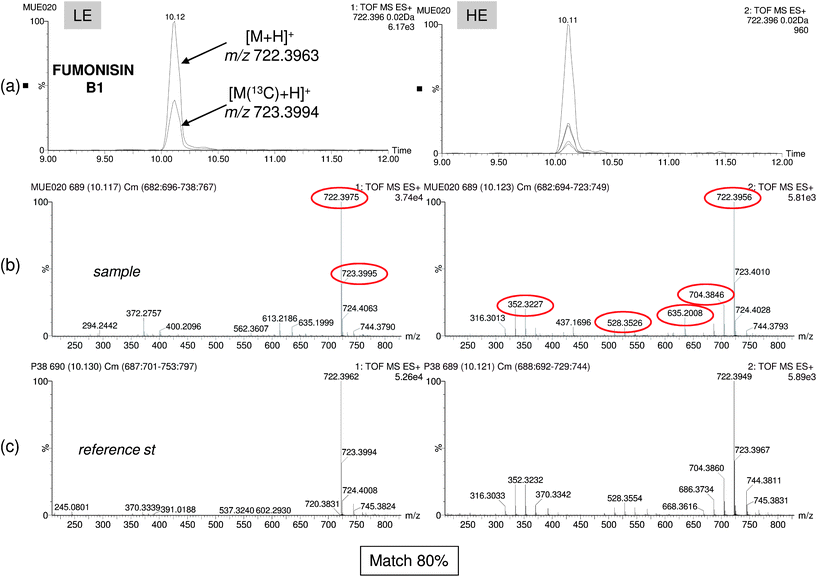 | ||
| Fig. 7 Non-target screening using experimental library search. Accurate-mass confirmation of the mycotoxin fumonisin B1. (a) Overlapped nw-XICs of the main deconvoluted ions of Fumonisin B1 under LE and HE conditions. Mass spectrum at LE and HE functions for sample (b) and reference standard (c). Library match (80%) and accurate-mass confirmation of the ions (mass errors below 1.3 mDa). | ||
When employing the theoretical, library-based, non-target screening approach, the investigation of findings when reference standards were unavailable was carried out using the same two strategies discussed before for post-target screening.
This work shows that the post-target approach has better capability for wide-scope screening of different analyte/sample matrix combinations. However, the non-target approach still has some advantages, especially when using an experimental library, as a comparison of the suspect compound versus library spectra is automatically performed achieving a highly reliable identification. In addition, other non-expected compounds that might be present in samples at relatively high concentrations might be detected without any kind of selection (pre- or post-target). However, searching for unknowns is an analytical challenge, where the possibilities to elucidate the components detected are rare.38 The main limitation for this approach is the difficulty of having large compound libraries similar to those used in GC-MS. At the moment, spectral libraries for LC-MS are home-made and are quite limited. Hopefully, in the near future large standardized libraries which will facilitate non-target screening will be available.
In this work, several contaminants have been found in the three types of samples investigated. Some of them have been tentatively identified without reference standard. The compounds detected belong to very different chemical classes and included emerging contaminants such as pharmaceuticals, UV filters and drugs of abuse, as well as several pesticides. Commonly used post-harvest fungicides imazalil and thiabendazol were identified in the orange and banana samples. In the case of the corn samples, the mycotoxins Fumonisin B1 and B2 were found, and also the less commonly detected Fumonisin B3 and B4 that were not previously included in the common pre-target approaches applied.
Conclusions
The comparison of different strategies based on the use of UHPLC coupled with QTOF MS for large-scale screening of organic pollutants in food, environmental and urine samples has been carried out. Thanks to the accurate-mass, full-spectrum acquisition in QTOF MS, it is feasible to apply both the target and non-target approaches, which can be seen as complementary within the public health field.The application of the target approach to selected samples has been demonstrated as an efficient tool for screening a large number of pollutants. For this purpose, a database containing information on the exact mass of the (de)protonated molecule and on the fragment ions and adducts (typically sodium adducts) has been created containing more than 1000 entries. This database has been built on the basis of our own experience and from data reported in the literature on LC-MS analysis of the compounds. Once a compound is detected, the potential positives need to be confirmed taking into account the information obtained on accurate masses of the (de)protonated molecule and of fragment ions, as well as the isotopic distribution. This is feasible using the MSE acquisition mode in the QTOF instrument, which allows the simultaneous MS data acquisition at low and high collision energy. The accomplishment of retention times and experimental MSE fragmentation using reference standards obviously facilitates the confirmation step.
In this work, an empirical library containing 231 selected compounds has also been employed in both the target and non-target approaches. Building empirical spectral libraries has been found to be the best way to facilitate both screening types, although it requires the injection of a large number of reference standards to be efficiently applied.
The non-targeted screening presents important drawbacks at low compound concentrations, especially in more complex-matrix samples, due to the difficulties in the components detection step. Identification of non-target contaminants is greatly facilitated when the compound detected is included in the home-made libraries, otherwise the elucidation of the compound becomes an analytical challenge where the possibilities of success are rare.
An interesting advantage associated with TOF MS-based methodologies concerns the possibility of performing retrospective analysis. This allows investigation of the presence of organic contaminants that were included in the first screening. This can be done at any time, without the need of either new analysis or new sample injections.
Acknowledgements
This work has been developed with financial support from the Ministry of Education and Science, Spain (CTQ 2009-12347). R. Diaz is very grateful to Conselleria d'Educació (Generalitat Valenciana) for his pre-doctoral grant. The authors are grateful to Serveis Centrals d'Instrumentació Científica (SCIC) of University Jaume I for the use of UPLC-QTOF-MS (QTOF Premier) and to Generalitat Valenciana for the financial support (Research Group of Excellence, Prometeo/2009/054).References
- F. Hernández, J. V. Sancho, M. Ibáñez and C. Guerrero, TrAC, Trends Anal. Chem., 2007, 26, 466 CrossRef.
- A. R. Fernández-Alba and J. F. García-Reyes, TrAC, Trends Anal. Chem., 2008, 27, 973 CrossRef.
- Y. Picó, C. Blasco and G. Font, Mass Spectrom. Rev., 2004, 23, 45 CrossRef.
- R. Rodil, J. B. Quintana, P. López-Manía, S. Muniategui-Lorenzo and D. Prada-Rodríguez, Anal. Chem., 2008, 80, 1307 CrossRef CAS.
- M. Petrovic, M. Gros and D. Barcelo, J. Chromatogr., A, 2006, 1124, 68 CrossRef CAS.
- E. Beltrán, M. Ibáñez, J. V. Sancho and F. Hernández, Rapid Commun. Mass Spectrom., 2009, 23, 1801 CrossRef.
- F. Hernández, J. V. Sancho, M. Ibáñez and S. Grimalt, TrAC, Trends Anal. Chem., 2008, 27, 862 CrossRef.
- A. Kaufmann, P. Butcher, K. Maden and M. Widmer, J. Chromatogr., A, 2008, 1194, 66 CrossRef CAS.
- A. Polettini, R. Gottardo, J. P. Pascali and F. Tagliaro, Anal. Chem., 2008, 80, 3050 CrossRef CAS.
- I. Ferrer and E. M. Thurman, J. Chromatogr., A, 2007, 1175, 24 CrossRef CAS.
- A. Kaufmann, P. Butcher, K. Maden and M. Widmer, Anal. Chim. Acta, 2007, 586, 13 CrossRef CAS.
- M. J. Martínez Bueno, A. Agüera, M. J. Gómez, M. D. Hernando, J. F. García-Reyes and A. R. Fernández-Alba, Anal. Chem., 2007, 79, 9372 CrossRef.
- C. A. Mueller, W. Weinmann, S. Dresen, A. Schreiber and M. Gergov, Rapid Commun. Mass Spectrom., 2005, 19, 1332 CrossRef CAS.
- A. C. Hogenboom, J. A. van Leerdam and P. de Voogt, J. Chromatogr., A, 2009, 1216, 510 CrossRef CAS.
- J. M. Marín, E. Gracia-Lor, J. V. Sancho, F. J. López and F. Hernández, J. Chromatogr., A, 2009, 1216, 1410 CrossRef.
- F. Hernández, O. J. Pozo, J. V. Sancho, L. Bijlsma, M. Barreda and E. Pitarch, J. Chromatogr., A, 2006, 1109, 242 CrossRef.
- B. Kmellár, P. Fodor, L. Pareja, C. Ferrer, M. A. Martínez-Uroz, A. Valverde and A. R. Fernandez-Alba, J. Chromatogr., A, 2008, 1215, 37 CrossRef.
- S. Inoue, T. Saito, H. Mase, Y. Suzuki, K. Takazawa, I. Yamamoto and S. Inokuchi, J. Pharm. Biomed. Anal., 2007, 44, 258 CrossRef CAS.
- L. Bijlsma, J. V. Sancho, E. Pitarch, M. Ibáñez and F. Hernández, J. Chromatogr., A, 2009, 1216, 3078 CrossRef CAS.
- M. Krauss, H. Singer and J. Hollender, Anal. Bioanal. Chem., 2010, 397, 943 CrossRef CAS.
- T. Pihlström, G. Blomkvist, P. Friman, U. Pagard and B. Österdahl, Anal. Bioanal. Chem., 2007, 389, 1773 CrossRef.
- C. Lesueur, P. Knittl, M. Gartner, A. Mentler and M. Fuerhacker, Food Control, 2008, 19, 906 CrossRef CAS.
- H. G. J. Mol, P. Plaza-Bolaños, P. Zomer, T. C. De Rijk, A. A. M. Stolker and P. P. J. Mulder, Anal. Chem., 2008, 80, 9450 CrossRef CAS.
- I. Ferrer, A. Fernandez-Alba, J. A. Zweigenbaum and E. M. Thurman, Rapid Commun. Mass Spectrom., 2006, 20, 3659 CrossRef CAS.
- Z. Herrera Rivera, E. Oosterink, L. Rietveld, F. Schoutsen and L. Stolker, Anal. Chim. Acta, 2011, 70, 114 CrossRef.
- M. Kellmann, H. Muenster, P. Zomer and H. Mol, J. Am. Soc. Mass Spectrom., 2009, 20, 1464 CrossRef CAS.
- I. Bobeldijk, J. P. C. Vissers, G. Kearney, H. Major and J. A. Van Leerdam, J. Chromatogr., A, 2001, 929, 63 CrossRef CAS.
- F. Hernández, S. Grimalt, O. J. Pozo and J. V. Sancho, J. Sep. Sci., 2009, 32, 2245 CrossRef.
- F. Hernández, M. Ibáñez, O. J. Pozo and J. V. Sancho, J. Mass Spectrom., 2008, 43, 173 CrossRef.
- S. Grimalt, O. J. Pozo, J. V. Sancho and F. Hernández, Anal. Chem., 2007, 79, 2833 CrossRef CAS.
- T. Portóles, M. Ibáñez, J. V. Sancho, F. J. López and F. Hernández, J. Agric. Food Chem., 2009, 57, 4079 CrossRef.
- P. Marquet, N. Venisse, E. Lacassie and G. Lachâtre, Analysis, 2000, 28, 925 CrossRef CAS.
- R. Díaz, M. Ibáñez, J. V. Sancho and F. Hernández, Rapid Commun. Mass Spectrom., 2011, 25, 355 CrossRef.
- F. Hernández, O. J. Pozo, J. V. Sancho, F. J. López, J. M. Marín and M. Ibáñez, TrAC, Trends Anal. Chem., 2005, 24, 596 CrossRef.
- M. Ibáñez, J. V. Sancho, F. Hernández, D. McMillan and R. Rao, TrAC, Trends Anal. Chem., 2008, 27, 481 CrossRef.
- O. J. Pozo, M. Barreda, J. V. Sancho, F. Hernández, J. Lliberia, M. A. Cortés and B. Bagó, Anal. Bioanal. Chem., 2007, 389, 1765 CrossRef CAS.
- European Commission Decision 2002/657/EC, Off. J. Eur. Communities: Legis., 2002, 221, 8 Search PubMed.
- M. Ibáñez, J. V. Sancho, O. J. Pozo, W. Niessen and F. Hernández, Rapid Commun. Mass Spectrom., 2005, 19, 169 CrossRef.
- F. Hernández, L. Bijlsma, J. V. Sancho, R. Díaz and M. Ibáñez, Anal. Chim. Acta, 2011, 684, 96 CrossRef.
- T. Portolés, E. Pitarch, F. J. López, J. V. Sancho and F. Hernández, J. Mass Spectrom., 2007, 42, 1175 CrossRef.
- M. Ibáñez, C. Guerrero, J. V. Sancho and F. Hernández, J. Chromatogr., A, 2009, 1216, 2529 CrossRef.
- O. J. Pozo, P. V. Eenoo, K. Deventer and F. T. Delbeke, TrAC, Trends Anal. Chem., 2008, 27, 657 CrossRef CAS.
- R. Oertel, N. Arenz, J. Pietsch and W. Kiroh, J. Sep. Sci., 2009, 32, 238 CrossRef CAS.
- B. Kasprzyk-Hordern, R. M. Dinsdale and A. J. Guwy, J. Chromatogr., A, 2007, 1161, 132 CrossRef CAS.
- A. Ojha, R. Rathod, C. Patel and H. Padh, Chromatographia, 2007, 66, 853 CAS.
- R. P. Shah, A. Sahu and S. Singh, J. Pharm. Biomed. Anal., 2010, 51, 1037 CrossRef CAS.
- G. N. W. Leung, D. K. K. Leung, T. S. M. Wan and C. H. F. Wong, J. Chromatogr., A, 2007, 1156, 271 CrossRef CAS.
- L. Kristoffersen, E. L. Øiestad, M. S. Opdal, M. Krogh, E. Lundanes and A. S. Christophersen, J. Chromatogr., B: Anal. Technol. Biomed. Life Sci., 2007, 850, 147 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c1ay05385j |
| This journal is © The Royal Society of Chemistry 2012 |