A simple in-house dry ashing chamber for the rapid determination of total mercury in organic-rich solid materials by oxidative pyrolysis followed by CVAAS and FI-ICPMS detection
M. V.
Balarama Krishna
*,
A. C.
Sahayam
and
D.
Karunasagar
National Center for Compositional Characterization of Materials (CCCM), Bhabha Atomic Research Centre, Department of Atomic Energy, Hyderabad. 500 062, India. E-mail: mvbalaramakrishna@rediffmail.com; Fax: +91 4027125463; Tel: +91 4027121365
First published on 30th November 2011
Abstract
A simple in-house pyrolysis chamber made of quartz material is described for the determination of total mercury content in a wide variety of organic-rich solid materials (plant, fish tissues and polymer based) by oxidative pyrolysis and cold vapour atomic absorption spectrometry (CVAAS). Oxidative pyrolysis was carried out by means of a Bunsen burner in the presence of an oxygen stream. Mercury liberated from the sample was collected in 0.1% KMnO4 trapping solution. Determination of mercury was achieved by CVAAS. Under optimal conditions, the limit of detection (LOD), calculated as the concentration of mercury yielding a signal equivalent to three times of the standard deviation of the blank value (3σ), obtained for CVAAS in conjunction with the oxidative pyrolysis method was found to be 0.08 ng g−1. Flow injection inductively coupled plasma mass spectrometry (FI-ICPMS) was used to validate the developed method. The results obtained for several reference materials, such as mussel tissue (CE-278), tuna fish (CE-463), tuna fish (464), polyethylene (EC-681), plankton (CRM-414), lichen (CRM-482) and human hair (CRM-397), by the developed method agreed well with the certified values. These studies clearly demonstrate the capabilities of the pyrolysis set-up that can potentially be used for the rapid determination of total mercury in a wide variety of organic-rich matrices.
Introduction
Mercury is widely considered as a global pollutant and is identified as a highly toxic element because of its accumulative and persistent character in the environment.1–4 Due to its neurological health impacts, mercury is one of the trace metals (As, Be, Cd, Co, Cr, Hg, Ni, Mn, Pb, Sb and Se) that the Environmental Protection Agency (EPA) has included for potential control under the Clean Air Act Amendments (U.S.EPA 2005).5 As a result of both natural and anthropogenic activities, mercury is among the most highly bio-concentrated toxic trace metals in various biological and environmental samples such as animal tissues, plant and coal from where it re-enters the human food chain.6–8Because of the “cold vapour” property of mercury, cold vapour atomic absorption spectrometry (CVAAS) has become the most widely used analytical technique employed for the total mercury determination in a great variety of environmental, biological and food samples.9–12Cold vapour atomic fluorescence spectrometry (CVAFS) has also gained a lot of popularity due to its better sensitivity in the last two decades.13–16 ICP-based spectrometric techniques such as inductively coupled plasma mass spectrometry (ICP-MS)17–19 and inductively coupled plasma optical emission spectrometry (ICP-OES)20–23 have also been used for mercury determinations. During the last four decades, many researchers have developed various approaches for the determination of mercury and its compounds.4,8,24,25 In general, closed microwave digestion with a variety of combinations of strong mineral acids and oxidants (e.g., H2O2) is routinely used for the decomposition/digestion of solid materials for the determination of mercury.26 But the microwave-assisted digestion procedures are limited by longer operation times (few hours), excessive pressure build-up (>500 psi) during the digestion process, and the potential loss of volatile elements, in particular, mercury.27
The most common sample introduction technique allowing solid sampling with ICP-MS is laser ablation (LA)28,29 and electrothermal vaporisation (ETV) for handling solid and slurried samples.30,31 However, with LA, accurate quantification is not an easy task in the absence of a reference material. Stability of the slurry, its homogeneity, particle size and sedimentation are some of the limitations of slurry sample preparation methods.25 Hence, direct solid analysis of mercury by means of GFAAS (relatively inexpensive and straightforward) and ETV-ICP-MS (multi-element, but more expensive) has not achieved much widespread use.
Most of the above mentioned limitations could be alleviated by using the dry ashing/pyrolysis method as an alternative to conventional wet digestion methods. Typical dry ashing temperatures are 450–800 °C at atmospheric pressure and the ash residues are dissolved in an appropriate acid for subsequent analysis. Several papers have been published about dry ashing as a sample preparation method for the determination of elemental composition data in a wide variety of matrices.25,32–34 In dry ashing the organic part of the sample is ashed under dry conditions to convert it into carbon (C) followed by formation of carbon monoxide and/or carbon dioxide. Hence the time required for complete volatilization of the matrix ranges from one hour to several hours depending on the type of matrix and the amount of sample material.32 However, the C oxidation step can be made rapid by using PGE (Pt, Pd and Rh) as catalysts or by purging with oxidising gases like oxygen.35
Various studies on the determination of mercury using the pyrolysis approach have also been reported in the literature.36–44 Melaku et al. have investigated the application of dry ashing for the determination of total mercury in several biological and environmental matrices.37 Young Park et al. have determined the mercury contents in various types of coal samples using acid extraction and pyrolysis methods.38 A commercially available mercury analyzer based on sample pyrolysis, gold amalgamation and AAS was evaluated for direct determination of total-Hg in various tobacco products.39
In the present study, a simplified and robust method was developed for the determination of total mercury using an in-house pyrolysis chamber and a KMnO4 solution as absorbent followed by CVAAS detection. The performance of the method has been compared with the FI-ICPMS technique to examine the interferences if any caused due to release of organic gaseous products during the pyrolysis process. Seven certified reference materials covering a wide range of matrices are used for the validation purposes. For optimization of the pyrolysis procedure, lichen BCR-CRM 482, fish homogenate ERM-CE 463 and polyethylene ERM-EC 681 were chosen as representatives of the three diverse matrices: plant-based, fish tissues and polymer-based, respectively.
Experimental
Instrumentation
Mercury was analyzed by cold vapour atomic absorption spectrometry (CV-AAS) at the 253.7 nm line using a mercury analyzer (Model MA 5800E, Electronics Corporation of India Ltd., Hyderabad, India).A VG Plasma Quad 3 ICP-MS (VG Elemental, Winsford, Cheshire, UK) fitted with a standard double Pass-Scott spray chamber and a concentric nebuliser was used for Hg detection. The mercury signal was monitored by the high abundant isotopes of mercury m/z 199, 200 and 202 using flow injection sample introduction mode. A simple FI system was assembled from a six-port injection valve (Cole-Parmer, UK) with a 100 μL sample loop. A time resolved mode of data acquisition (TRA) was used for obtaining FI-ICPMS plots. The sensitivity of the instrument was checked each day before starting the experiments. The optimized ICP-MS operating conditions that yielded the best sensitivity for mercury are summarized in Table 1.
| Instrumental parameters | Scanning parameters |
|---|---|
| Coolant gas: 13.4 L min−1 | Scanning mode: peak jump |
| Aux. gas: 0.97 L min−1 | Number of replicates: 3 |
| Nebulizer gas: 0.85 L min−1 | Dwell time: 300 μs per channel |
| Sampler cone: 1.0 mm Ni | Sample delay: 30 s |
| Skimmer cone: 0.7 mm Ni | Stabilization delay: 20 s |
| Torch type: Fassel | Flow rate: 1 mL min−1 |
| Plasma FW power: 1350 W | Sample injection loop volume: 100 μL |
| Reflected power: <5 W | Isotopes monitored: m/z 199, 200, 202 |
Standards, reagents and materials
All solutions were prepared in ultra-pure water (>18 MΩ cm resistivity), obtained from a Milli-Q high purity water system, located in a class 100 area. Analytical reagent grade chemicals were used without further purification. Sub-boiled HCl and HNO3 were prepared in-house by sub-boiling distillation in quartz stills. Mercury standard solution (1000 mg L−1) in 5% HNO3 (traceable to NIST 3133) (SD Fine-Chem Ltd, Mumbai, India) was used as a stock standard. The calibration solutions for Hg in the range of 1–100 ng mL−1 were prepared by sequential dilution of a 1 mg L−1 stock standard solution with ultra-pure water. All the stock standard solutions were stored in a refrigerator at 4 °C. A 2% sodiumborohydride (NaBH4) (Merck, Darmstadt, Germany) (m/v) solution was prepared freshly daily by dissolving the appropriate amount of solid in 0.3% NaOH solution (m/v) and was used as reducing agent. 10% HCl solution was used as a carrier solution during mercury measurements with CVAAS. Potassium permanganate solution was prepared by dissolving an appropriate amount of the salt in high purity water and used as a trapping solution for Hg.The homemade pyrolysis chamber was a hollow cylinder (10 cm × 5 cm i.d.) made of quartz fused to a B24 joint. The other end of the chamber was fused to a 10 cm × 1 cm i.d. quartz tube. A small tube was projected inside the quartz chamber as shown in Fig. 1 to insert a thermocouple to monitor the temperature inside the chamber during the pyrolysis process.
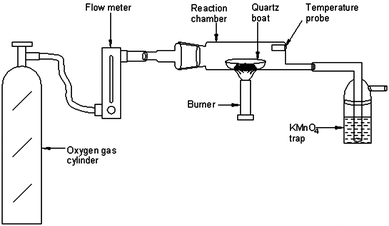 | ||
| Fig. 1 Schematic diagram of the pyrolysis set-up for the determination of total mercury in organic-rich solid materials. | ||
All containers were soaked in 20% HNO3 and cleaned thoroughly with high purity water prior to use. The certified reference materials (CRMs) used in this study are from European Reference Materials (ERM)-CE-278 Mussel Tissue, CE-463, 464 Tuna fish, EC-681 Polyethylene and from Community Bureau of Reference (BCR), CRM-414 Plankton, CRM-482 Lichen and CRM-397 Human hair.
General analytical procedure for total-Hg determination
A schematic diagram of the pyrolysis system for mercury release from a wide variety of organic-rich matrices is depicted in Fig. 1. It comprises a flow meter connected to an oxygen cylinder, a homemade pyrolysis chamber and a KMnO4 solution for trapping of mercury vapour. For optimization of the pyrolysis procedure, lichen CRM 482, fish homogenate CE 463 and polyethylene-EC 681 were chosen as representatives of the three diverse matrices: plant-based, fish tissues and polymer-based respectively.Accurately weighed amounts (∼100 mg) of these representative samples were placed in the quartz boat and then inserted into the pyrolysis chamber which was then heated to ∼550 °C with a Bunsen burner. The oxygen gas flow rate was maintained at 300 mL min−1. The two main parts, the pyrolysis chamber and the trapping solution cell, are linked with short segments of silicone tubing. After the pyrolysis step, the KMnO4 solution (pink colour) was decomposed on adding HCl followed by gentle heating at 85–90 °C on an IR hot-plate (Schott Instruments, Germany). The heating process was continued until the pink colour of the solution completely disappeared. At the end of the experiment, the samples were made up to the required volume with high purity water for analysis by CVAAS using 1 mL of 2% NaBH4 as the reducing agent. For FI experiments with ICP-MS, the sample was loaded into a 100 μL loop (load mode) and injected into the 2% HCl carrier solution (inject mode) at a flow rate of 1 mL min−1 and the plots were obtained for each aliquot of sample solutions. Peak area measurements were used for quantification of mercury in processed sample solutions. All the experiments were run in triplicate.
Process blank solutions were also prepared in the same way without taking any sample material in the quartz boat. To know the loss of mercury, if any, during decomposition of KMnO4 solution and also for quantification of mercury in processed samples, matrix matched standards were prepared by spiking a known amount of mercury in the KMnO4 solution (keeping the KMnO4 concentration the same as the absorbent solution) before and after decomposition. Standard blank solutions were also prepared using only KMnO4 solution which were then decomposed as above. The optimized pyrolysis procedure was applied to certified values of the reference materials.
Results and discussion
In order to evaluate the analytical applicability of the proposed pyrolysis method to diverse matrices using an in-house pyrolysis chamber for the determination of mercury, three representative reference materials of different origins (plant, fish tissues and polymer based) were used for optimization experiments. The volume of the KMnO4 trapping solution was fixed at 5 mL in all the studies carried out in this work.Pyrolysis system optimization
Direct determination of Hg in organic-rich matrices by CV-AAS (without any pre-treatment procedures) is complicated by the presence of organic compounds. Therefore, it is necessary to decompose the matrix and separate the interfering organic products prior to mercury determination. Hence the present pyrolysis approach was developed which can significantly remove such kinds of interferences and simplify the quantification of mercury in the samples of complex nature.As described in the earlier section, the gaseous mercury atoms/or species produced in the pyrolysis process are likely to originate from pyrolytic reduction of (in the presence of carbon) bound Hg ions in the sample. Based on this principle, it was observed that various parameters such as pyrolysis temperature, sample heating time, oxygen flow rate, sample mass and concentration of KMnO4 in the trapping solution are primarily responsible for quantitative recovery of mercury from solid materials. Hence detailed studies were carried out to examine their influence.
Pyrolysis temperature
As mentioned before, typical dry ashing temperatures are 450–800 °C at atmospheric pressure. But the main objective of the proposed pyrolysis approach is its analytical applicability to diverse matrices. Hence, the effect of temperature of the pyrolysis chamber (varied from 300 to 650 °C) on the recovery of mercury was studied with the three representative materials using the general procedure by keeping the sample heating time, concentration of KMnO4 absorbent solution and sample mass fixed at 3 min, 0.1% and 100 mg respectively. In Fig. 2, the effect of pyrolysis temperature on the recovery of mercury is shown for the representative samples tested here. It is interesting to observe that for the polyethylene sample, quantitative recovery (>95%) of mercury was achieved for pyrolysis temperatures of ∼400 °C while it requires >500 °C for the plant and fish tissue materials. Variations in the pyrolysis temperature between the lichen, tuna fish and polyethylene materials may possibly be due to their difference in matrix composition. It was also found that no memory effects of mercury exist when the temperature was ∼550 °C. Hence a pyrolysis temperature of ∼550 °C was chosen for further studies.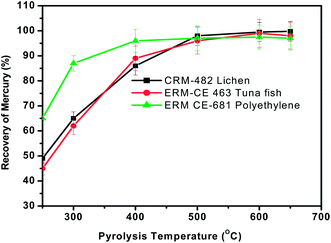 | ||
| Fig. 2 Effect of pyrolysis temperature on the recovery of total mercury from the three different representative samples. Sample heating time = 3 min, oxygen gas flow rate = 300 mL min−1 and concentration of KMnO4 trapping solution = 0.1%. | ||
Sample heating time
As discussed earlier, sample heating in the presence of oxygen promotes the complete oxidation of the matrix and release of mercury from solid materials. Hence the effect of heating time on the recovery of mercury was assessed between 1 min and 5 min by keeping the sample mass (∼100 mg), carrier gas flow rate (300 mL min−1) and concentration of trapping solution (0.1%) constant. The sample was heated in a Bunsen burner in the presence of a continuous oxygen stream until the evolution of smoke was ceased. The pyrolysis efficiency at each time interval was calculated by measuring the mercury content in the KMnO4 trap solution and comparing the value with the certified value of that particular material. The % recovery of Hg obtained at different time intervals is shown in Fig. 3. As seen from Fig. 3, >70% recovery of Hg was obtained within one min of sample heating but quantitative recovery of mercury (>95%) was achieved with a sample heating time of 3 min and was used for further studies.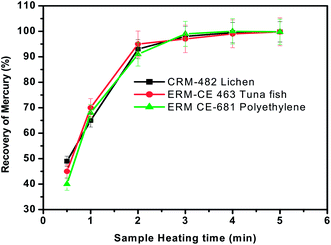 | ||
| Fig. 3 Effect of sample heating time on the recovery of total mercury from the three different representative samples. Oxygen gas flow rate = 300 mL min−1 and concentration of KMnO4 trapping solution = 0.1%. | ||
Geng et al. had developed the oxygen flask combustion (OFC) method for the determination of Hg in ash and soil samples.40 But the time required for the analysis of each sample was >30 min while the pyrolysis approach in the present work typically required <6 min to complete a total mercury analysis including the pyrolysis process (3–4 min), decomposition of KMnO4 solution (<2 min) and CVAAS analysis (∼1 min).
Oxygen gas flow rate (carrier gas)
The presence of oxygen is essential for the rapid oxidation of the organic matter present in the tested materials. Therefore, the influence of oxygen flow rate on the recovery of mercury was investigated with all the three representative materials using the general procedure by keeping the sample heating time, concentration of KMnO4 absorbent solution and sample mass fixed at 3 min, 0.1% and 100 mg respectively. The results obtained when the oxygen flow rate was varied from 50 mL min−1 to 500 mL min−1 (controlled by a rotameter) are presented in Fig. 4. As seen here, the recovery of mercury was quantitative when the oxygen flow rate was >200 mL min−1. The reason for low recovery of mercury is not very clear when the oxygen flow rate is <200 mL min−1, but this behaviour is likely to be caused by a combination of factors such as incomplete combustion due to shortage of oxidant and deposition of mercury at cooler regions of the pyrolysis chamber. Further studies would be needed to pinpoint the exact cause of such discrepancies.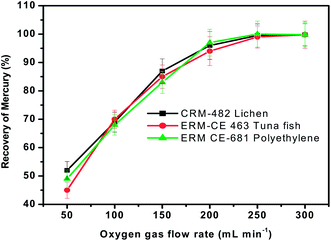 | ||
| Fig. 4 Influence of oxygen gas flow rate on the recovery of total mercury from three different representative samples. Sample heating time = 3 min and concentration of KMnO4 trapping solution = 0.1%. | ||
Effect of sample mass
It is possible that the emission of heavy smoke (gaseous pyrolysis products) from these organic-rich matrices during the pyrolysis process may influence the quantitative recovery of mercury if large sample amounts are used. Hence the effect of the mass of sample introduced for pyrolysis with the existing set-up (as shown in Fig. 1) on the recovery of mercury must be assessed. To study this effect, the proposed pyrolysis method was carried out using different masses of reference materials ranging from 50 to 300 mg at a heating temperature of ∼550 °C, heating time of 3 min and oxygen gas flow of 300 mL min−1. The general procedure as described previously was followed. Fig. 5 shows the effect of sample mass on mercury recovery, where it is evident that, for a given sample mass up to 200 mg, quantitative recovery (>97%) could be achieved for all the three representative samples. As the sample mass increased (>200 mg), the recovery of mercury decreased from 97% to 90%. It was generally observed that a large amount of thick fumes were generated during pyrolysis when the mass of sample is sufficiently high (>200 mg). Due to this, the interaction of mercury vapour with the KMnO4 solution might be greatly reduced and hence mercury vapours simply exit from the trap solution along with other gaseous products. This feature may be responsible for the low recovery of mercury as the sample mass increases. From these studies, it was observed that the sample masses of up to 0.2 g were suitable for the quantitative determination of Hg by this approach.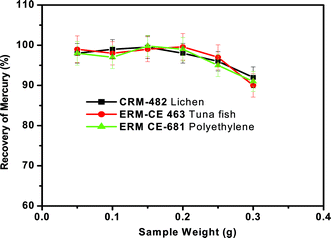 | ||
| Fig. 5 Effect of concentration of KMnO4 trapping solution on the recovery of total mercury from the three different representative samples. Oxygen gas flow rate = 300 mL min−1 and sample heating time = 3 min. | ||
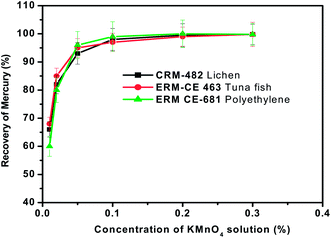
Concentration of KMnO4 trapping solution
Various trapping solutions such as acidified KMnO4, K2Cr2O7, liquid nitrogen cooled-trap, acidified KI solution and aqueous bromine solution have been reported in the literature for the absorption of gaseous mercury species.45–47 In the present studies, acidified KMnO4 solution (0.1% KMnO4–3% HCl) was chosen as the reagent for trapping of mercury due to the ease of preparation and its high trapping efficiency.The relation between the trapping efficiency for mercury and the concentration of KMnO4 in the trapping solution in the range of 0.01 to 0.2% was investigated by keeping the sample weight (∼100 mg), pyrolysis time (3 min), oxygen gas flow rate (300 mL min−1) and volume of the KMnO4 trapping solution (5 mL) constant. Then the general pyrolysis procedure for the release of mercury was followed. The experimental observations are presented in Fig. 6. Results of the experiments showed that if the concentration of KMnO4 in the trapping solution was more than 0.1%, the efficiency always remained close to 100%. But when the concentration of KMnO4 was less than 0.1%, the trapping efficiency declined as the concentration of KMnO4 decreased. If a very dilute solution (<0.01%) is used, the solution lost the pink colour of KMnO4 during the sampling procedure. It may be that mercury vapour generated during pyrolysis was oxidized by KMnO4 and then remained in the solution. As a compromise, the concentration of KMnO4 was fixed at 0.1% for mercury trapping purposes in all the subsequent pyrolysis experiments carried out in the present work.
 | ||
| Fig. 6 Effect of the mass of sample on the recovery of total mercury from the three different representative samples. Oxygen gas flow rate = 300 mL min−1, sample heating time = 3 min and concentration of KMnO4 trapping solution = 0.1%. | ||
Although the trapping efficiency of KMnO4 is excellent (∼100%), it severely suppresses the absorption signal during mercury measurements by CVAAS due to its strong oxidizing capabilities and also it consumes a major portion of the reducing agent which is used for the generation of elemental Hg prior to CVAAS measurements. Therefore, it is necessary to decompose the excess KMnO4 prior to mercury determination. Various decomposition methods such as addition of hydroxylamine hydrochloride and oxalic acid under warm conditions have been reported in the literature.40 In the present study, HCl was used for this purpose as it can also serve as a carrier acid during CVAAS measurements. After the pyrolysis process, 0.5 mL of HCl was added to the KMnO4 trap solution and heated gently on an IR hot-plate by maintaining the temperature at about 85–90 °C. The heating process was continued until the pink colour of the solution completely disappeared. This whole process takes less than 2 min.
In conclusion, the optimum conditions are as follows: sample mass <200 mg, oxygen gas flow rate 300 mL min−1, sample heating time 3 min and concentration of KMnO4 trapping solution 0.1%.
Analytical performance
The analytical performance of the proposed oxidative pyrolysis method was studied by examining the three parameters namely limit of detection (LOD), recovery of mercury and precision (relative standard deviation (RSD)).The calibration curves for both aqueous mercury standards and mercury spiked KMnO4 solutions (before and after decomposition) were generated for the quantification of mercury. The first set of sample solutions were prepared by spiking known amounts of mercury standard (2.5, 5 and 10 ng mL−1) in 5 mL of 0.1% KMnO4 solution followed by decomposition in the presence of HCl as described in the earlier section. In another set of sample solutions, three different aliquots of 0.1% KMnO4 (5 mL each) were initially decomposed by the addition of conc. HCl and then spiked the same amounts of mercury standard (2.5, 5 and 10 ng mL−1) as in the case of first set of sample solutions. The process blank solutions were also prepared in the same manner without adding mercury standard solution. The FI-ICPMS experiments for both standard blank and process blank solutions showed no trace amounts of mercury present in KMnO4 solution.
The calibration plots of FI-ICPMS obtained with the first and second set of solutions with the 100 μL injection loop are shown in Fig. 7. In all the cases, the mercury concentration was determined by taking peak area measurements. The calibration plots (Fig. 7) obtained with spiked mercury standards show that the recovery of Hg was well above 98%. This study clearly indicated that there was no significant loss of mercury during the KMnO4 decomposition process. As shown in Table 2, the analytical curves for both aqueous and KMnO4 trapping solutions exhibit almost the same slope and good reproducibility. This allows the use of aqueous standard calibration for quantification purposes.
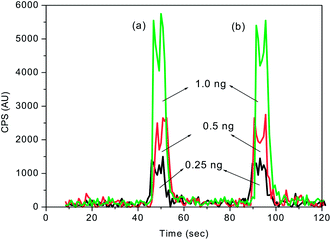 | ||
| Fig. 7 FI-ICPMS calibration plots obtained for different concentrations of mercury spiked (a) before and (b) after decomposition of 0.1% KMnO4 trapping solution. Sample injection volume = 100 μL. | ||
| Medium | Response function | R 2 | ||
|---|---|---|---|---|
| CVAAS | FI-ICPMS | CVAAS | FI-ICPMS | |
| a Calibration range: 1–100 ng mL−1. | ||||
| Aqueous | y = 0.002x + 0.001 | y = 4074x − 854.2 | 0.999 | 0.999 |
| KMnO4 (mercury spiked before decomposition) | y = 0.001x + 0.002 | y = 3979x + 270.5 | 0.997 | 0.999 |
| KMnO4 (mercury spiked after decomposition) | y = 0.001x + 0.001 | y = 4110x + 312.2 | 0.998 | 0.999 |
Under optimal conditions, the limit of detection (LOD) (three times the standard deviation of the blank solutions) obtained for CVAAS in conjunction with the oxidative pyrolysis method was found to be 0.08 ng g−1. The RSD values obtained from the three replicate determinations of the seven reference materials taken through the general pyrolysis procedure were in the range of 1–8%. The accuracy of the method was established by analyzing seven certified reference materials of different origin. The mercury values obtained for these samples as listed in Table 3 are in close agreement with their respective certified values. Meanwhile, the recovery of the mercury values was found to be quantitative with the values ranging from 96 to 102% (Table 3), indicating the absence of (a) any significant losses of mercury and (b) matrix interference or interference due to gaseous pyrolysis products.
| Type of reference material | Certified value of mercury/μg g−1 | Obtained in this work/μg g−1 | Recovery of mercury (%) | ||
|---|---|---|---|---|---|
| CVAAS | FI-ICPMS | CVAAS | FI-ICPMS | ||
| European Reference Materials (ERM) | |||||
| Mussel Tissue ERM-CE 278 | 0.196 ± 0.009 | 0.192 ± 0.007 | 0.193 ± 0.009 | 96.9 | 98.5 |
| Tuna Fish ERM-CE 463 | 2.85 ± 0.16 | 2.89 ± 0.13 | 2.83 ± 0.18 | 101.4 | 99.3 |
| Tuna Fish ERM-CE 464 | 5.24 ± 0.10 | 5.17 ± 0.11 | 5.21 ± 0.09 | 98.7 | 99.4 |
| Polyethylene ERM-EC 681 | 4.5 ± 0.15 | 4.61 ± 0.17 | 4.33 ± 0.14 | 102.2 | 96.2 |
| Community Bureau of Reference (BCR) | |||||
| Plankton BCR-CRM-414 | 0.276 ± 0.018 | 0.272 ± 0.014 | 0.269 ± 0.016 | 98.4 | 97.5 |
| Lichen BCR-CRM-482 | 0.48 ± 0.02 | 0.46 ± 0.02 | 0.47 ± 0.03 | 96.5 | 98.6 |
| Human Hair BCR-CRM-397 | 12.30 ± 0.50 | 12.47 ± 0.43 | 12.26 ± 0.15 | 101.3 | 99.7 |
As discussed in the earlier sections, the released organic pyrolysis products such as C, CO and CO2 may be trapped in KMnO4 absorbent solution along with the lines of mercury during the oxidative pyrolysis decomposition process. Hence the proposed pyrolysis method was validated using the FI-ICPMS method to examine the potential interferences of organic pyrolysis products that are likely to be trapped in KMnO4 solution. But, excellent agreements were obtained from inter-method (CVAAS and FI-ICPMS) comparisons of reference materials of different matrices tested in the present work (Table 3). These observations clearly indicate that mercury vapours were selectively trapped in the KMnO4 solution and the remaining pyrolysis products are simply coming out of the absorbent solution, thus completely eliminating the interferences in AAS measurements. A typical plot obtained for the lichen representative reference material taken through the complete pyrolysis process and FI-ICPMS analysis is shown in Fig. 8. The three peaks in Fig. 8 correspond to different aliquots of the lichen material. These studies clearly indicate that the Hg determinations in highly complex and organic-rich materials were highly reproducible with the present pyrolysis set-up.
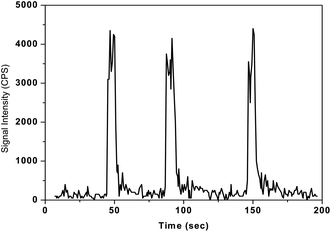 | ||
| Fig. 8 Typical plot showing the reproducibility of the developed pyrolysis-FI-ICPMS method for three successive aliquots of lichen-482 reference material. Sample mass = 100 mg, sample heating time = 3 min, concentration of KMnO4 solution = 0.1%, sample injection volume = 100 μL and final volume of the sample solution = 5 mL. | ||
Conclusions
A simple oxidative pyrolysis—CVAAS—method has been developed using an in-house pyrolysis chamber for the determination of total mercury in a wide variety of organic-rich solid materials. Inter-comparison experiments with FI-ICPMS clearly indicated that the developed pyrolysis approach significantly removes interferences due to gaseous pyrolysis products and simplifies the quantification of mercury in the samples of complex nature. The results obtained by the developed method were in good agreement with certified values. The relative standard deviation (RSD) was better than 8% for most of the tested materials. The proposed pyrolysis method offers the advantages of the absence of sample digestion, reduced reagents, and thus helps in obtaining accurate results in the ultra-trace analysis, particularly for the determination of mercury. It also has inherent advantages including relatively low cost instrumentation, minimal operating expenses and ease of use. The method also has a rapid turn-around time, with each sample taking about 6 minutes. As no catalysts need to be employed, problems of contamination and exhaustion of the catalyst cartridges are eliminated. Thus the proposed oxidative pyrolysis—CVAAS—method may be employed economically for producing quality results for mercury in organic-rich samples.Acknowledgements
The authors are thankful to Dr J. Arunachalam, Head, CCCM, and Dr Sunil Jai Kumar, Head, Ultra-Trace Analysis Section, CCCM, for their constant support and encouragement.References
- W. L. Clevenger, B. W. Smith and J. D. Winefordner, Crit. Rev. Anal. Chem., 1997, 27, 1 CrossRef CAS
.
- T. W. Clarkson, Crit. Rev. Clin. Lab. Sci., 1997, 34, 369–403 CrossRef CAS
- J. Brent, Clin. Toxicol., 2001, 39(7), 707–710 CrossRef CAS
- M. Leemakers, W. Baeyens, P. Quevauviller and M. Horvat, TrAC, Trends Anal. Chem., 2005, 24(5), 383–393 CrossRef
- U.S. EPA, Standards of performance for new and existing stationary sources: electric utility steam generating units, Fed. Regist., 2005, 70, 28605 Search PubMed
- K. A. Francesconi, Analyst, 2007, 132, 17–20 RSC
- M. Horvat and R. D. Wilken, Fresenius J. Anal. Chem., 2000, 366, 415 CrossRef CAS
- J. E. Sanchez Uria and A. Sanz-Medel, Talanta, 1998, 47, 509 CrossRef CAS
- M. A. Vieira, A. S. Ribeiro, A. J. Curtius and R. E. Sturgeon, Anal. Bioanal. Chem., 2007, 38(4), 837–847 CrossRef
- S. Rio Segade and J. F. Tyson, Spectrochim. Acta, Part B, 2003, 58, 797–807 CrossRef
- M. V. Balarama Krishna, M. Ranjit, D. Karunasagar and J. Arunachalam, Talanta, 2005, 67, 70–80 CrossRef
- D. P. Torres, D.L. G. Borges, V. L. A. Frescura and A. J. Curtius, J. Anal. At. Spectrom., 2009, 24, 1118–1122 RSC
- L. Ebdon, M. F. Foulkes, S. Le Roux and R. Munoz-Olivas, Analyst, 2002, 127(8), 1108–1114 RSC
- P. B. Stockwell, W. T. Corns and J. Allen, J. Anal. At. Spectrom., 2009, 24, 1026–1033 RSC
- D. Sanchez-Rodas, W. T. Corns, B. Chen and P. B. Stockwell, J. Anal. At. Spectrom., 2010, 25, 933–946 RSC
- Y. Gao, W. Yang, Ch. Zheng, X. Hou and Li Wu, J. Anal. At. Spectrom., 2011, 26, 126–132 RSC
- D. Beauchemin, Anal. Chem., 2010, 82, 4786–4810 CrossRef CAS
- M. V. Balarama Krishna, K. Chandrasekaran and D. Karunasagar, Talanta, 2010, 81, 462–472 CrossRef CAS
- M. V. Balarama Krishna, G. Venkateswarlu, A. K. Sanjukta and D. Karunasagar, At. Spectrosc., 2011, 32(4), 127–144 Search PubMed
- I. Serafimovski, I. Karadjova, T. Stafilov and J. Cvetkovic, Microchem. J., 2008, 89, 42–47 CrossRef CAS
- X. Zhu and S. D. Alexandratos, Microchem. J., 2007, 86, 37–41 CrossRef CAS
- E. J. dos Santos, A. B. Herrmann, F. Vieira, C. S. Sato, Q. B. Correa, T. A. Maranhao, L. Tormen and A. J. Curtius, Microchem. J., 2010, 96(1), 27–31 CrossRef CAS
- J. Castro, J.C. Spraul and R. Kenneth Marcus, Anal. Methods, 2009, 1, 188–194 RSC
- S. Diez and J. M. Bayona, Talanta, 2008, 77, 21–27 CrossRef CAS
- M.d. G. A. Korn, E. S. da Boa Morte, D. C. M. B. dos Santos, J. T. Castro, J. T. P. Barbosa, A. P. Teixeira, A. P. Fernandes, B. Welz, W. P. C. dos Santos, E. B. G. N. dos Santos and M. Korn, Appl. Spectrosc. Rev., 2008, 43, 67–92 CrossRef CAS
- K. J. Lamble and S. J. Hill, Analyst, 1998, 123, 103R–133R RSC
-
Introduction to Microwave Sample Preparation, in Theory and Practice, ACS Professional Reference Book, ed. H. M. Kingston and L. B. Jassie, American Chemical Society, Washington DC, 1988 Search PubMed
- V. L. Dressler, D. Pozebon, M. F. Mesko, A. Matusch, U. Kumtabtim, B. Wu and J. Sabine Becker, Talanta, 2010, 82(5), 1770–1777 CrossRef CAS
- T. Stehrer, J. Heitz, J. D. Pedarnig, N. Huber, B. Aeschlimann, D. Günther, H. Scherndl, T. Linsmeyer, H. Wolfmeir and E. Arenholz, Anal. Bioanal. Chem., 2010, 398(1), 415–424 CrossRef CAS
- I. Gelaude, R. Dams, M. Resano, F. Vanhaecke and L. Moens, Anal. Chem., 2002, 74(15), 3833–3842 CrossRef CAS
- M. Resano, I. Gelaude, R. Adams and F. Vanhaecke, Spectrochim. Acta, Part B, 2005, 60, 319–326 CrossRef
- S. Demirel, M. Tuzen, S. Saracoglu and M. Soylak, J. Hazard. Mater., 2008, 52, 1020–1026 CrossRef
- C. S. Kira and V. A. Maihara, Food Chem., 2007, 100, 390–395 CrossRef CAS
- A. C. Sahayam, G. Venkateswarlu and S. C. Chaurasia, At. Spectrosc., 2009, 30(4), 139–142 CAS
- A. C. Sahayam, S. C. Chaurasia and G. Venkateswarlu, Anal. Chim. Acta., 2010, 661, 17–19 CrossRef CAS
- R. Dumarey and R. Dams, Microchim. Acta, (Wien), 1984, III/304, 191–198 Search PubMed
- A. Melaku, I. Gelaude, F. Vanhaecke, L. Moens and R. Dams, Microchim. Acta, 2003, 142, 7–12 CrossRef
- J. Y. Park, J. H. Wo and T. G. Lee, Energy Fuels, 2006, 20, 2413–2416 CrossRef CAS
- Y. M. Panta, S. Qian, C.L. Cross and J. V. Cizdziel, J. Anal. Appl. Pyrolysis, 2008, 83, 7–11 CrossRef CAS
- W. Geng, T. Nakajima, H. Takanashi and A. Ohki, J. Hazard. Mater., 2008, 154, 325–330 CrossRef CAS
- C. T. Costley, K. F. Mossop, R. JohnDean, L. M. Garden, J. Marshall and J. Carroll, Anal. Chim. Acta, 2000, 405, 179–183 CrossRef CAS
- R.-J. Huang, Z.-X. Zhuang, Y. Tai, R.-F. Huang, X.-R. Wang and F. S. C. Lee, Talanta, 2006, 68, 728–734 CrossRef CAS
- C. Bin, W. Xiaoru and F. S. C. Lee, Anal. Chim. Acta, 2001, 447, 161–169 CrossRef
- J. M. Ombaba, Microchem. J., 1996, 53, 195–200 CrossRef CAS
- L. Ebdon and J. R. Wilkinson, Analyst, 1982, 107, 269–275 RSC
- Z. Wang and S. O. Pehkonen, Atmos. Environ., 2004, 38, 3675–3688 CrossRef CAS
- P. Worathanakul, P. Kongkachuichay, J. D. Noel, A. Suriyawong, D. E. Giammar and P. Biswas, Environ. Eng. Sci., 2008, 25(7), 1061–1070 CrossRef CAS
| This journal is © The Royal Society of Chemistry 2012 |