Measuring P2X1 receptor activity in washed platelets in the absence of exogenous apyrase†
Kari B.
Anderson
,
Welivitiya
Karunarathne
and
Dana M.
Spence
*
Michigan State University, Department of Chemistry, East Lansing, MI 48824, USA. E-mail: dspence@chemistry.msu.edu; Tel: 517.355.9715 x174
First published on 7th November 2011
Abstract
Purinergic receptor
signaling events in platelets are a major determinant in platelet function. However, investigating the ATP-sensitive P2X1 platelet receptor is difficult due to its rapid desensitization in the washed platelet sample matrix. To minimize desensitization, most studies involving P2X1 activity in washed platelets require apyrase in the sample to reduce matrix ATP levels. Unfortunately, the apyrase will also rapidly degrade any ATP added exogenously during the studies. Here, we describe a method that employs the reported P2X1 inhibitor NF449 to sensitize washed platelets in the absence of any added apyrase. Sensitization is verified by spectrofluorometric determination of Ca2+ entry into the platelets after stimulation with concentrations of ATP ranging from 0.625 μM to 5 μM. Results suggest that sensitization of the P2X1 receptor by NF449 is not necessarily dependent upon the inhibitor concentration, but rather the ratio of the inhibitor to exogenously-added ATP concentrations. With a ratio of ATP agonist to NF440 concentration of ∼5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, the resulting percent change in fluorescence due to Ca2+ entry into the platelet is 39.3 ± 0.8%; however, at a ratio of 1:8 ATP to NF449 the percent change is reduced to 13.1 ± 2.2%. The sensitizing effect is also investigated as a function of time. The results obtained verify that NF449 can behave as a concentration-dependent inhibitor and sensitizer of the plateletP2X1 receptor in washed platelet samples, depending on the ATP concentration in the matrix.
:1, the resulting percent change in fluorescence due to Ca2+ entry into the platelet is 39.3 ± 0.8%; however, at a ratio of 1:8 ATP to NF449 the percent change is reduced to 13.1 ± 2.2%. The sensitizing effect is also investigated as a function of time. The results obtained verify that NF449 can behave as a concentration-dependent inhibitor and sensitizer of the plateletP2X1 receptor in washed platelet samples, depending on the ATP concentration in the matrix.
Introduction
The primary function of platelets in the bloodstream involves maintaining hemostasis and preventing blood loss through the clotting process. Upon endothelial cell injury, von Willebrand factor and sub-endothelial collagen are exposed resulting in platelet activation.1,2Adenine receptors on the surface of the platelet are important in this process, most notably the ADP-sensitive receptors P2Y1/P2Y12, and the ATP-sensitive P2X1 receptor.3–6Though roles for P2Y-type receptors in platelet activation and thrombus formation are well established, the role of the P2X1 receptor in platelet function is less clear. For example, activation of the P2X1 receptor by ATP has been reported to be responsible for extracellular Ca2+ entry into the platelet. This P2X1 activation has been shown to induce transient platelet shape change,7 and is also involved with collagen-induced aggregation events.8–11 It has also been shown that P2X1, along with ATP, is important as a secondary platelet agonist in the activation process.12 Conversely, other studies report a minimal role for P2X1 in platelet activation.13 Not only is Ca2+ important for P2X1 response, but also ADP dependent P2Y response where extracellular Ca2+ dependent ectonucleotidases affect ADP induced platelet aggregation.14 Finally, a recent review on the P2X1 receptor stresses its importance in thrombotic events and the need to study the receptor as a therapeutic target.15
The P2X1 receptor is difficult to study because it is rapidly desensitized during in vitro experimentation, especially when the platelets have been separated from plasma and washed.16,17 In order to overcome this rapid receptor desensitization,18–20apyrase, which catalyzes the dephosphorylation of adenine nucleotides, is commonly used during platelet purification from whole blood to reduce the number of phosphate-containing nucleotides already present in the bulk solution. Unfortunately, the addition of apyrase to the bulk solution results in the breakdown of exogenously added ATP to ADP, which is unable to activate the P2X1 receptor.
Another factor contributing to the complexity of plateletP2X1 activation is distinguishing effects due to ATP from those arising from ADP, especially after dephosphorylation from ATP to ADP. For example, when P2X1 is stimulated with ATP, Ca2+ enters the platelet from the sample matrix and can be measured with a fluorescent probe such as Fura-2 or Fluo4-AM. However, ADP can also contribute to this Ca2+ signal, as ADP binding to the P2Y1 receptor results in the release of Ca2+ from granules contained within the platelet.21–23 Therefore, the degradation of ATP to ADP, or ADP contamination in the ATP agonist, leads to inconclusive results regarding the source of the increased platelet Ca2+. However, there are techniques that can be used to distinguish the two receptors. For example, in the case of measuring Ca2+, one can simply perform the studies in a Ca2+-free buffer. Such a system would prevent Ca2+ entering the platelet due to P2X1 activation, but not P2Y activation (because the Ca2+ release due to P2Y activation is from granules already in the platelet).
In addition to using Ca2+-free buffers to differentiate receptor activity, many investigators employ the use of α,β-methylene-ATP (α,β-me-ATP), a stable ATP analogue that cannot be enzymatically dephosphorylated to ADP. The use of such a stable ATP analogue ensures that any measured Ca2+ flux into the platelet is due to P2X1. However, α,β-me-ATP leads only to transient platelet shape change7 and low levels of aggregation24 and could potentially be masking true P2X1 activity that would be measured with authentic ATP.
To facilitate the understanding of receptor action and response, inhibitors of the platelet purinergic receptors can be employed. One of the most widely-used plateletP2X1 inhibitors is NF449, which has been shown to reduce Ca2+ influx and reduce activation of the receptor by both ATP and α,β-me-ATP.25–27 Interestingly, in contrast to its inhibitory actions on the P2X1 receptor, NF449 has been reported to enhance P2X1 sensitization in frog oocytes.27 A detailed examination of these reports involving NF449 inhibition and sensitization reveals that the conditions (concentrations of NF449, number of cells used in the studies and incubation time of the NF449 with the cells prior to measurement) differed greatly. These contrasting reports led to the hypothesis that NF449 may exhibit sensitizing and inhibitory actions on the plateletP2X1 receptor that are dependent upon the ratio of agonist and inhibitor employed during platelet preparation.
Experimental
Isolation of platelets from human whole blood
Whole blood was obtained via venipuncture under consent from donors and collected into heparinized tubes. In general, 10 tubes of whole blood were collected from each donor (∼80 mL). After the whole blood was centrifuged at 500 g for 10 min, the platelet-rich plasma (PRP) was decanted for subsequent platelet purification.Platelets were isolated from the PRP by adding 1 mL of acid citrate dextrose (ACD; in mM, 41.6 citric acid anhydrous, 76.7 sodium citric acid anhydrous, 122.1 dextrose) to every 9 mL of PRP and centrifuging at 1500 g for 10 min. The harvested platelets were then washed twice with 10% ACD calcium free Modified Tyrodes Buffer (MTB; in mM, 12 NaHCO3, 0.32 NaH2PO4, 10 HEPES, 137 NaCl, 2.7 KCl, 0.5 MgCl2, and 5.5 dextrose) solution with centrifugation in between each wash. The washed platelets were then re-suspended in 10% ACD calcium free MTB. No apyrase was used in this purification. The washed platelets were counted using a hemacytometer and adjusted to a concentration of 2.0 × 109platelets mL−1 in 1 mL 10% ACD calcium free MTB. Platelet samples were prepared and investigated the same day as isolation.
Fluo 4 AM probe and NF449 platelet sample preparation
A 2.3 mM stock of the calcium specific probe Fluo 4 AM (Molecular Probes/Invitrogen Eugene, OR) was prepared by dissolving 50 μg of the probe in 20 μL of anhydrous DMSO. A 5 μL aliquot of the stock was mixed with 5 μL of a 200 mg mL−1pluronicF-127 surfactant (Molecular Probes/Invitrogen Eugene, OR) solution to enhance the probe's penetration capacity. Finally, this probe solution was diluted to 1 mL in 10% ACD calcium free MTB to create an 11.4 μM working solution. This 1 mL solution of the fluorescent probe was then added to the 1 mL platelet solution for a final concentration of 5.7 μM Fluo 4 AM and incubated on ice for 30 min. The platelet solution containing the probe was then centrifuged at 1500 g for 5 min and washed twice with 10% ACD calcium free MTB to remove any excess probe. Finally, the platelets were resuspended in 2 mL of 10% ACD calcium free MTB.Aliquots of the Fluo 4 AM loaded platelet solution (1.0 × 109platelets mL−1) were incubated for 30 min on ice with NF449 (Tocris Bioscience, Ellisville, MO) at final concentrations ranging from 0–40 μM.
Fluorescence determination of intracellular platelet Ca2+
Calcium measurements were performed with a spectrofluorometer (HORIBA Jobin Yvon, Eison, NJ) having 2 nm slit widths, excitation at 496 nm and emission at 516 nm, along with a 21N-Q-10 far UV quartz cuvette. A 40 μL aliquot of platelet sample was added to 1940 μL of MTB with calcium (2 mM Ca) at 37 °C and a fluorescence baseline was collected for 15 s prior to the addition of 20 μL of agonist prepared in Ca Free MTB. Agonists included ATP, ADP and α,β-methylene ATP (Sigma-Aldrich, St. Louis, MO) at stock concentrations ranging from 0–1000 μM resulting in final concentrations of 0–10 μM in the cuvette. Fluorescence measurements were collected for an additional 90 s; samples were stirred throughout the measurement. A schematic of sample preparation and subsequent fluorescence measurement is shown in Fig. 1.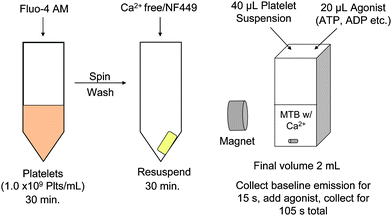 | ||
| Fig. 1 Schematic representation of experimental setup. Platelets were incubated with the Ca2+ probe Fluo-4 AM for 30 mins, then the excess probe was washed off and the platelets incubated with either buffer or NF449. Finally, measurements of Ca2+ influx were performed using a spectrofluorometer with a magnet setup so that samples could be stirred during measurement. Samples were kept in a Ca2+ free environment until measurement. | ||
Data analysis
Included in the results and discussions section are spectra that represent raw fluorescence data collected over time, whereas the summarized data is represented by a percent change in fluorescence. This percent change was calculated by subtracting the quotient of the average baseline fluorescence intensity and average peak fluorescence intensity from 1 and multiplying by 100 to obtain a percent.Results and discussions
NF449 sensitizes ATP-induced Ca2+ influx
ATP was added as an agonist to platelets, in the presence and absence of NF449, to evaluate its effect on P2X1 mediated Ca2+ influx. Shown in Fig. 2 (left), adding CaMTB to platelets alone has no effect on the fluorescence signal (trace A). Furthermore, the addition of ATP to the platelet sample resulted in no significant increase in fluorescence signal (compared to baseline) due to Ca2+ entry into the platelets (trace B). However, there was a significant increase in Ca2+ entry when ATP was added to platelets containing 0.5 μM NF449 (trace C). Additionally, as shown in Fig. 2 (right), Ca2+ entry into the platelets containing 2.5 μM NF449 increased as the concentration of ATP was increased from 0.625 μM to 5 μM, although the first measurable change in signal (in comparison to baseline) did not occur until the ATP concentration reached 310 nM (data for lower ATP concentrations not shown for figure clarity). Thus, activation of the P2X1 receptor and subsequent Ca2+ influx is ATP concentration dependent under NF449 sensitizing conditions.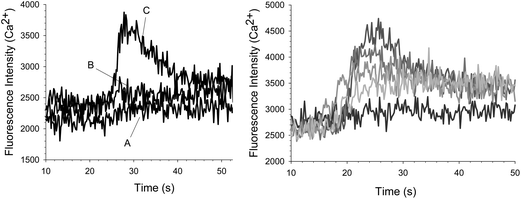 | ||
| Fig. 2 (Left) Fluorescence measurements taken over time for platelets in the presence and absence of NF449. No significant increase in Ca2+ is measured when CaMTB (trace A) or 2.5 uM ATP (trace B) is added to platelets alone. However, an increase in Ca2+ was measured when 2.5 uM ATP was added to platelets containing 0.5 uM NF449 (trace B). (Right) The effect of ATP on platelet Ca2+ influx with a constant concentration of 2.5 uM NF449. As the concentration of ATP was increased from 0.625 μM, to 1.25 μM, to 2.5 μM up to 5 μM, the intracellular platelet Ca2+ also increased as compared to baseline where no ATP was added. | ||
Though it would be beneficial to study ATP and NF449 effect with dose response curves, the curve would be heavily influenced by NF449 concentration as well as platelet count. Therefore, any EC50/IC50 data gleaned from this would only be relevant to these particular experimental parameters. Furthermore, this method could be utilized to study ATP binding to the P2X1 receptor since the only study that produces a Kd for this interaction uses α,β-me-ATP, not native ATP.28
Effect of Ca2+ free buffer on Ca2+ entry into the plateletviaATP and ADP agonists
A possible explanation for the increase in fluorescence emission shown in Fig. 2 is that the ATP contained ADP impurities that were stimulating P2Y-type receptors, resulting in the release of Ca2+ stores in the platelet granules. Therefore, studies were also performed in a Ca2+-free buffer; in this construct, activation of the P2X1 receptor with ATP would not result in an increase of fluorescence emission due to Ca2+ entry into the platelet because there is no Ca2+ in the buffer to cross the platelet membrane. The data in Fig. 3 (left) show that ATP addition to platelets containing NF449 in a Ca2+ free environment result in no increase in fluorescence signal (trace B). The top trace in Fig. 3 (left) is another aliquot of platelets from the same sample. However, this trace was obtained using a Ca2+-containing buffer and, as shown, Ca2+ entry into the platelet was detected (trace A). Finally, when ADP was added to NF449-containing platelets, there was an increase in fluorescence signal (trace C) even in the absence of Ca2+, which is likely due to Ca2+ release from granular stores in the platelets. Collectively, the summarized data in Fig. 3 (right) suggests that the ATP induced Ca2+ entry into NF449-containing platelets is through P2X1 activation and not solely a result of an ADP impurity contained in the ATP agonist.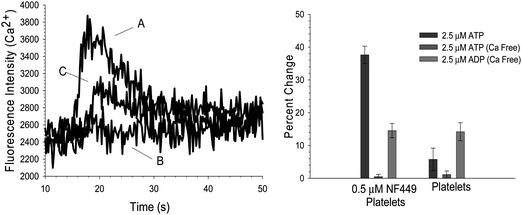 | ||
| Fig. 3 Effect of Ca2+ free buffer on Ca2+ influx in platelet containing 0.5 μM NF449. (Left) 2.5 μM ATP was added as an agonist in the presence of Ca2+ as represented in trace A. The typical increase in Ca2+ influx is seen. However, in a Ca2+ free environment (trace B), the addition of 2.5 μM ATP resulted in no significant increase in Ca2+. Additionally, in the Ca2+ free environment, ADP is still able to elicit a Ca2+ increase (trace C). (Right) The summarized data indicates that when Ca2+ is not present, there is no Ca2+ influx due to 2.5 μM ATP in platelets containing 0.5 μM NF449 or platelets alone. However, signal due to ADP stimulation is still present. | ||
Effect of α,β-me-ATP concentrations on Ca2+ entry into the platelet
The P2X1 receptor is most commonly studied using a form of ATP that is stable, such as α,β-me-ATP. This form of ATP ensures that breakdown to ADP does not occur, thus minimizing effects from P2Y-type events. Fig. 4 shows a Ca2+ influx due to the addition of α,β-me-ATP (trace B) to NF449 containing platelets (18.7 ± 0.9%), a significantly weaker signal (P < 0.001) in comparison to authentic ATP (trace A) stimulation (39.3 ± 0.8%). No calcium influx was seen when ATP or α,β-me-ATP were added to platelets alone (traces C and D). Unlike ATP, higher concentrations of α,β-me-ATP did not elicit calcium influx (data not shown), suggesting the potential for α,β-me-ATP to desensitize the P2X1 receptor more readily than ATP itself.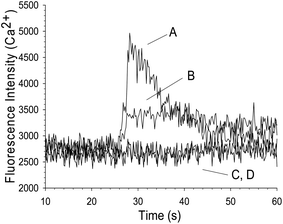 | ||
| Fig. 4 Effect of α,β-me-ATP on platelet Ca2+ influx. Both A and B traces indicate the presence of platelets containing 0.5 μM NF449, but the agonist for trace A is 2.5 μM ATP and the agonist for trace B is 2.5 μM α,β-me-ATP. Increases in Ca2+ influx were measured in the case of both agonists, but the increase due to ATP was larger than the increase due to α,β-me-ATP. The two controls in this figure, represented as C, D, are platelets alone that were treated with both agonists, neither showed an increase in Ca2+ influx. | ||
Effect of NF449 concentrations on Ca2+ entry into the platelet
NF449 is generally regarded as a competitive inhibitor of the P2X1 receptor and at concentrations greater than 1 μM, will have non-specific inhibitory effects on P2Y receptors. However, data in Fig. 1–3 suggest receptor sensitization at a concentration of 0.5 and 2.5 μM NF449. Therefore, it was anticipated that concentrations of NF449 less than 1 μM would also have the same sensitizing effects. Fig. 5 summarizes the effect of varying NF449 concentrations from 62.5 nM up to 40 μM in the presence of 2.5 μM ATP as the agonist for P2X1 stimulation and subsequent Ca2+ entry into the platelet. As shown, the signal resulting from the fluorescence emission due to Ca2+ entry into the platelet begins to increase with increments of NF449. However, as the concentration of NF449 continues to increase, an inhibitory effect emerges (for an ATP concentration of 2.5 μM), resulting in reduced calcium entry into the platelet.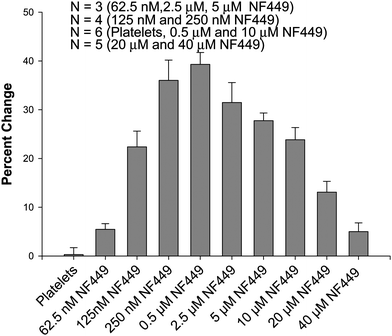 | ||
| Fig. 5 Effect of NF449 concentration on platelet Ca2+ influx due to 2.5 μM ATP. As NF449 concentration is increased from 0 to 0.5 μM, the Ca2+ influx increases, but as the concentration of NF449 is further increased from 0.5 μM to 40 μM the Ca2+ influx decreases from the measured maximum. | ||
Importantly, when the NF449 concentrations reached inhibitory levels (of the 2.5 μM ATP), the addition of higher concentrations of ATP were able to restore the Ca2+ entry into the platelets. For example, at an NF449 concentration of 0.5 μM, the percent change in fluorescence emission upon the addition of 2.5 μM ATP was 39.3 ± 2.4%. This value dropped to 13.1 ± 2.2% when the NF449 concentration was increased to 20 μM. However, when the ATP concentration was raised to 10 μM, the percent change in fluorescence emission recovered to a value of 34.1 ± 2.8%. Sample spectra are shown in Figure S1. These results suggest that the ratio between ATP and NF449 is key in stimulating or inhibiting Ca2+ increase through the P2X1 receptor. Therefore, if the ATP concentration in a particular study is 2.5 μM, the optimum sensitizing concentration of NF449 is ∼0.5 μM, while inhibitory concentrations of NF449 would be obtained at concentrations >10 μM. Of course, these values of NF449 would change if the ATP concentrations were changed.
Time dependence of NF449 sensitization of P2X1
Previously, NF449 has been shown to sensitize P2X1, although the effect was measured at picomolar concentrations on single oocytes and only lasted for a few seconds. In studies reported here, nanomolar to low micromolar concentrations exhibited this same sensitization feature even though the NF449 was allowed to incubate with the platelets for 30 min prior to the addition of agonist. Therefore, a study was performed to determine if the ability of NF449 to sensitize the plateletP2X1 receptor was time dependent. The time study was executed using platelets incubated with 62.5 nM NF449 followed by the addition of 2.5 μM ATP as the agonist. The results of this study, shown in Fig. 6, indicate that the fluorescence signal from Ca2+ entry due to addition of the ATP agonist decreased with time. The increase at 1 min is thought to be less than that of the 5 min signal due to NF449 not having enough time to diffuse to the platelet surface. The subsequent decrease in signal suggests that the sensitizing effect of NF449 is indeed time dependent, consistent with the previous report.27 However, it appears that increasing the NF449 concentration prolongs its sensitization abilities.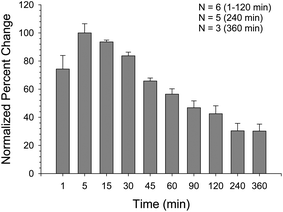 | ||
| Fig. 6 NF449 potency over time with 2.5 μM ATP agonist. Using 62.5 nM NF449, platelet Ca2+ influx was measured over the course of 6 h with the data being normalized to the 5 min. percent change in signal. The Ca2+ influx peaked at 5 min and decreased steadily over time. | ||
Conclusions
The P2X1 platelet receptor is difficult to study due to its rapid desensitization, especially when the platelets have been purified from plasma and reside in aqueous buffers void of any naturally occurring adenine nucleotidases. Therefore, in washed platelet samples, apyrase is typically added exogenously which, unfortunately, can also rapidly degrade any ATP added to the sample. Here, we provide evidence that the addition of NF449, a substance generally regarded to inhibit the plateletP2X1 receptor, has the ability to sensitize this receptor. Furthermore, the data shown in figure S2 suggest that NF449 is a more effective P2X1 sensitizer than apyrase. However, proper sample handling is key to this sensitizing ability as the concentration ratio of NF449 to ATP must be considered, as does the elapsed time before a measurement is made (as the sensitizing effect dissipates depending on the concentration of NF449 employed).References
- J. J. Sixma, G. H. van Zanten, E. U. M. Saelman, M. Verkleij, H. Lankhof, H. K. Nieuwenhuis and P. G. de Groot, Thromb. Haemostasis, 1995, 74, 454–459 CAS.
- V. T. Turitto, H. J. Weiss, T. S. Zimmerman and I. I. Sussman, Blood, 1985, 65, 823–831 CAS.
- S. P. Kunapuli, Z. Ding, R. T. Dorsam, S. Kim, S. Murugappan and T. M. Quinton, Curr. Pharm. Des., 2003, 9, 2303–2316 CrossRef CAS.
- M. P. Mahaut-Smith, S. J. Ennion, M. G. Rolf and R. J. Evans, Br. J. Pharmacol., 2000, 131, 108–114 CrossRef CAS.
- S. Murugappan and S. P. Kunapuli, Front. Biosci., 2006, 11, 1977–1986 CrossRef CAS.
- C. Vial, B. Hechler, C. Leon, J. P. Cazenave and C. Gachet, Thromb. Haemostasis, 1997, 78, 1500–1504 CAS.
- M. G. Rolf, C. A. Brearley and M. P. Mahaut-Smith, Thromb. Haemostasis, 2001, 85, 303–308 CAS.
- C. Y. E. Fung, C. A. Brearley, R. W. Farndale and M. P. Mahaut-Smith, Thromb. Haemostasis, 2005, 94, 37–40 CAS.
- B. Hechler, N. Lenain, P. Marchese, C. Vial, V. Heim, M. Freund, J.-P. Cazenave, M. Cattaneo, Z. M. Ruggeri, R. Evans and C. Gachet, J. Exp. Med., 2003, 198, 661–667 CrossRef CAS.
- M. P. Mahaut-Smith, G. Tolhurst and R. J. Evans, Platelets, 2004, 15, 131–144 CrossRef CAS.
- C. Oury, E. Toth-Zsamboki, C. Thys, J. Tytgat, J. Vermylen and M. F. Hoylaerts, Thromb. Haemostasis, 2001, 86, 1264–1271 CAS.
- C. Y. E. Fung, C. Cendana, R. W. Farndale and M. P. Mahaut-Smith, J. Thromb. Haemostasis, 2007, 5, 910–917 CrossRef CAS.
- S. Takano, J. Kimura, I. Matsuoka and T. Ono, Eur. J. Pharmacol., 1999, 372, 305–309 CrossRef CAS.
- S. Jones, R. J. Evans and M. P. Mahaut-Smith, Br J Haematol, 153, pp. 83–91 Search PubMed.
- M. P. Mahaut-Smith, S. Jones and R. J. Evans, Purinergic Signal Search PubMed.
- S. Valera, N. Hussy, R. J. Evans, N. Adami, R. A. North, A. Suprenant and G. Buell, Nature, 1994, 371, 516–519 CrossRef CAS.
- C. J. Lewis and R. J. Evans, Br. J. Pharmacol., 2000, 131, 1659–1666 CrossRef CAS.
- A. Baurand, A. Eckly, N. Bari, C. Leon, B. Hechler, J.-P. Cazenave and C. Gachet, Thromb. Haemostasis, 2000, 84, 484–491 CAS.
- C. Gachet, Thromb. Haemostasis, 2008, 99, 466–472 CAS.
- A. R. Hardy, P. B. Conley, J. Luo, J. L. Benovic, A. W. Poole and S. J. Mundell, Blood, 2005, 105, 3552–3560 CrossRef CAS.
- J. Jin and S. P. Kunapuli, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 8070–8074 CrossRef CAS.
- P. Savi, P. Beauverger, C. Labouret, M. Delfaud, V. Salel, M. Kaghad and J. M. Herbert, FEBS Lett., 1998, 422, 291–295 CrossRef CAS.
- C. Leon, B. Hechler, M. Freund, A. Eckly, C. Vial, P. Ohlmann, A. Dierich, M. LeMeur, J.-P. Cazenave and C. Gachet, J. Clin. Invest., 1999, 104, 1731–1737 CrossRef CAS.
- J. A. Erhardt, K. Pillarisetti and J. R. Toomey, J. Thromb. Haemostasis, 2003, 1, 2626–2635 CrossRef CAS.
- B. Hechler, S. Magnenat, M. L. Zighetti, M. U. Kassack, H. Ullmann, J.-P. Cazenave, R. Evans, M. Cattaneo and C. Gachet, J. Pharmacol. Exp. Ther., 2005, 314, 232–243 CrossRef CAS.
- M. Hohenegger, M. Waldhoer, W. Beindl, B. Boing, A. Kreimeyer, P. Nickel, C. Nanoff and M. Freissmuth, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 346–351 CrossRef CAS.
- M. Hulsmann, P. Nickel, M. Kassack, G. Schmalzing, G. Lambrecht and F. Markwardt, Eur. J. Pharmacol., 2003, 470, 1–7 CrossRef CAS.
- P. Savi, J. Bornia, V. Salel, M. Delfaud and J.-M. Herbert, Br. J. Haematol., 1997, 98, 880–886 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c1ay05530e |
| This journal is © The Royal Society of Chemistry 2012 |