Integrated DNA extraction and amplification using electrokinetic pumping in a microfluidic device
Joseph
Parton
,
Christopher
Birch
,
Cordula
Kemp
,
Stephen J.
Haswell
,
Nicole
Pamme
and
Kirsty J.
Shaw
*
Department of Chemistry, University of Hull, Cottingham Road, Hull, HU6 7RX, UK. E-mail: k.j.shaw@hull.ac.uk; Fax: +44 (0)1482 466416; Tel: +44 (0)1482 466746
First published on 3rd November 2011
Abstract
An integrated system employing anion exchange for the extraction of DNA from biological samples prior to polymerase chain reaction DNA amplification has been developed, based on microfluidic methodology utilising electrokinetic pumping. In this system, the biological samples were added directly to chitosan-coated silica beads to facilitate DNA immobilisation. The purified, pre-concentrated DNA was then eluted using a combination of electro-osmotic flow enhanced with electrophoretic mobility, which enable DNA to be transported by both mechanisms into the DNA amplification chamber. Through optimisation of the DNA elution conditions, average DNA extraction efficiencies of 69.1% were achievable. Subsequent DNA amplification performed on the microfluidic system demonstrated not only the ability to use electrokinetic movement to integrate the two processes on a single device, but also that the quality and quantity of DNA eluted was suitable for downstream analysis. This work offers an attractive real-world to chip interface and a route to simpler Lab-on-a-Chip technology which eliminates the need for moving parts.
Introduction
Microfluidics has been widely exploited for genetic analysis in order to advance developments in areas such as clinical diagnostics and forensics.1,2 Across this wide range of potential applications, it is often necessary to first isolate the nucleic acids from the sample matrix using an extraction step. This allows the target DNA to be purified and/or pre-concentrated.At present, DNA extraction methodologies employed in microfluidic systems are predominantly silica-based, in which the addition of chaotropic salts to the sample matrix generates the necessary conditions for cell lysis and subsequent denaturing of the free DNA. This in turn leads to the binding of the DNA to a high surface area, solid silica phase.3,4 Any biological contaminants or potential inhibitors of downstream processes, such as the polymerase chain reaction (PCR), can then be removed using an alcohol wash and the purified DNA can be eluted in a low ionic strength buffer. While such methodologies have consistently provided a high yield of purified DNA, chaotropic salts and organic solvents have been found to substantially hinder the PCR process. As a result, attempts to integrate silica-based nucleic acid extraction to downstream processes on a single microfluidic device has carried with it the added complication of additional wash steps. In order to experimentally simplify the extraction procedure, modified materials such as movable magnetic silica particles, which carry the bound target DNA to the elution buffer rather than introducing sample and reagents consecutively, have been reported.5
The use of anion exchange resins presents an attractive alternative to silica-based methodologies as they bypass the need for chaotropic salts and organic solvents by utilising surface chemistries which facilitate binding and elution of DNA to/from the solid-phase matrix by pH manipulation. Nakagawa et al. developed a microfluidic device which integrated a silicon wafer coated with amino groups to allow DNA recoveries of 40% from whole blood by the capture and release of DNA at pH 7.5 and 10.6 respectively.6 More recently, the field has capitalised on a similar surface activity provided by the increasingly popular and highly versatile biomaterial chitosan (α(1 → 4)-linked 2-amino-2-deoxy-β-D-glucopyranose). The bioactive polymer, synthesised from the partial deacetylation of naturally occuring chitin, possesses a variety of physical characteristics, such as high solubility, viscosity and biodegradability.7 Importantly, the presence of reactive amino side groups with a pKa of 6.3 makes chitosan an ideal solid-phase matrix for DNA extraction as deprotonation, and hence DNA release, can occur at pH 9 leaving the DNA in a suitable media for subsequent PCR. Furthermore, the ability to treat silica surfaces with chitosan directly has inspired a number of microfluidic applications and several high performing DNA purification methodologies have emerged. Cao et al., for example, were able to integrate chitosan-coated beads into a microfluidic device and obtained DNA recoveries from whole blood as high as 75%.8 Simple hydrodynamic pumping techniques were implemented to flow sample and reagents through the bead-packed extraction chamber allowing easy retrieval of target DNA for further analysis. Reedy et al. used a similar approach in conjunction with a preceding silica-based step, highlighting chitosan as an especially effective tool for nucleic acid preconcentration. This additional step provided a significant 50-fold reduction in elution volume which is highly appealing as it is more manageable for PCR.9 In further work, Reedy and colleagues adopted a modified approach by using pre-fabricated polymeric micro-posts functionalised with chitosan. Also using hydrodynamic pumping, the work demonstrated the ability to functionalise a wider variety of materials (e.g.poly(methyl methacrylate)) with chitosan for DNA extraction.10
The use of electrokinetic movement of reagents, i.e. electro-osmotic flow (EOF) and/or electrophoresis, in microfluidic devices is a desirable alternative to hydrodynamic pumping, as it eliminates the need for moving parts and simplifies the experimental process both mechanically and spatially. Such techniques have been used to successfully integrate DNA extraction and amplification steps, demonstrating the process as a suitable real-world to chip interface.11 In addition to this, Shaw et al. showed how a reversal of normal EOF (i.e. using a net positive charge on the surface generating bulk flow towards the positive electrode) using a suitable surface functional group (e.g.hexadimethrine bromide) is especially welcome in analysis of nucleic acids, as the negatively-charged sugar-phosphate backbone ensures an electrophoretic movement which compliments the direction of the bulk flow. This combined electrokinetic movement can be advantageous, in terms of both experimental timescale and energy usage.11
In the work presented here, an integrated microfluidic device for performing DNA extraction and amplification is described in which all sample and reagent movement within the system is controlled electrokinetically. For the first time, the processes support an anion exchange methodology using chitosan-coated silica beads for the initial DNA purification step. Furthermore, the observed reverse-EOF in the presence of chitosan-treated surfaces is capitalised on for optimum experimental performance.
Experimental
Manufacture and functionalisation of microfluidic devices
Glass microfluidic devices were produced using standard photolithography and wet-etching techniques to produce the design shown in Fig. 1. The 1 mm base plate was etched to a depth of 100 μm and then thermally bonded to a 3 mm top plate containing 3 mm access ports. Silanisation of the PCR chamber was performed in order to prevent DNA polymerase adsorption which would otherwise lead to PCR inhibition.12 This was achieved by adding a 150 mM solution of trichloro(1H,1H,2H,2H-perfluorooctyl)silane [Sigma-Aldrich, UK] in 2,2,4-trimethylpentane [Fisher Scientific, UK] to the PCR chamber for 10 min. Solutions of 2,2,4-trimethylpentane, acetone and distilled water were then sequentially used to wash the device.13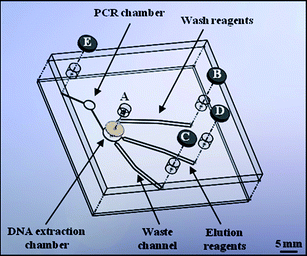 | ||
| Fig. 1 Schematic of the microfluidic device design used to perform integrated DNA extraction and amplification experiments. The position of the DNA extraction chamber packed with chitosan-coated silica beads, DNA amplification chamber and electrodes (B–E) for performing electrokinetic movement are all illustrated. | ||
Silica beads (40–60 μm diameter) [Sigma-Aldrich, UK] were thoroughly cleaned using piranha solution (2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, H2SO4/H2O2), washed with water and dried prior to use. The beads were then incubated with a solution of 1% low molecular weight chitosan oligosaccharide lactate [Sigma-Aldrich, UK] and 0.1% (3-glycidyloxypropyl) trimethoxysilane [Sigma-Aldrich, UK] for 8 h at room temperature. After chitosan coating, the beads were washed in 10 mM acetic acid and dried at 60 °C prior to use.8 When required, the chitosan-coated beads were injected through Port A into the DNA extraction chamber on the microfluidic device, which was designed such that the beads were held in place via the keystone effect.
:1, H2SO4/H2O2), washed with water and dried prior to use. The beads were then incubated with a solution of 1% low molecular weight chitosan oligosaccharide lactate [Sigma-Aldrich, UK] and 0.1% (3-glycidyloxypropyl) trimethoxysilane [Sigma-Aldrich, UK] for 8 h at room temperature. After chitosan coating, the beads were washed in 10 mM acetic acid and dried at 60 °C prior to use.8 When required, the chitosan-coated beads were injected through Port A into the DNA extraction chamber on the microfluidic device, which was designed such that the beads were held in place via the keystone effect.
DNA extraction and amplification procedure
In order to enable calculation of the DNA extraction efficiency of the system, human genomic DNA samples were prepared to a final concentration of 5 ng μl−1 in an artificial urine matrix.14 To further evaluate the system real urine samples were used during the integrated DNA extraction and amplification phase. The urine samples were thermally lysed and 50 μL aliquots used for analysis. The biological samples were then added to 10 mM 2-(N-morpholino)ethanesulfonic acid (MES) buffer (pH 5) prior to manual injection through Port A onto the solid-phase matrix in order to facilitate DNA binding, with any residual sample directed into the waste channel. Once the biological sample had been loaded, the microfluidic device was sealed with carbon-filled polystyrene electrodes positioned within the ports of the device and connected to an external Paragon 3B Power Supply Unit [Kingfield Electronics, UK] in order to facilitate electrokinetic movement.Following this, the solid-phase was washed with 10 mM MES buffer, contained within channel B, in order to remove any potential contaminants of downstream processes. This was achieved by applying a voltage between electrodes B and C. The pre-concentrated, purified DNA was then eluted using 10 mM Tris buffer with 50 mM KCl (pH 9), contained within channel D, by applying a voltage between electrodes D and E. Whilst optimising the electrokinetic movement it was found that cooling the microfluidic device to 4 °C increased the efficiency of the process by reducing Joule heating, which in turn minimized sample evaporation/diffusion.
Subsequently DNA amplificationvia PCR was performed on the microfluidic device. The PCR solution consisted of: 1x GoTaq®buffer, 2 mM MgCl2, 1 unit GoTaq® Hot Start DNA polymerase [Promega, UK], 10 mg ml−1 bovine serum albumin [NEB Inc., UK], 0.01% (w/v) poly(vinylpyrrolidone), 0.1% (v/v) Tween-20 [Sigma-Aldrich, UK], 200 μM each deoxyribonucleotide triphosphates [Bioline, UK] and 0.1 μM Amelogenin forward and reverse primers15 [Eurofins MWG Operon, Germany]. Thermal cycling was performed using a thermoelectric Peltier element, which provided both the heating and cooling required. The following program was used: an initial denaturation step of 95 °C for 2 min, 35 cycles of 94 °C for 30 s, 60 °C for 30 s and 72 °C for 30 s, with a final extension step of 60 °C for 7 min. Control PCR samples were also run on a Techne TC-312 thermal cycler.
DNA quantification and capillary electrophoresis
DNA quantification was performed using a Quant-iT™ PicoGreen® double stranded DNA Assay Kit [Invitrogen, UK] and analysed using a FLUOstar Optima Plate Reader [BMG Labtech, UK]. All amplified DNA samples were analysed off-chip by capillary gel electrophoresis using a 3500 Genetic Analyzer [Applied Biosystems, UK]. Samples (1 μL) were added to 12 μL of Hi-Di™ Formamide and 0.5 μL GeneScan™ 500 LIZ® Size Standard [Applied Biosystems, UK] and denatured for 5 min at 95 °C before being snap-cooled on ice and loaded onto the instrument.Results and discussion
Movement of reagents within the microfluidic device
Initial experiments were designed to assess the compatibility of the different reagents required for chitosan-based anion exchange methodology with EOF movement. It was found that all solutions were capable of supporting EOF and so a range of voltages (50–500 V cm−1) were examined to evaluate flow rates. The EOF mobilities for the wash and elution buffers were found to be 3.70 × 10−4 cm2 V−1sec−1 and 3.95 × 10−4 cm2 V−1sec−1, respectively (Fig. 2).![Graph showing the average νEOF at different applied voltages for the binding/wash solution [MES buffer pH 5 (■)] and elution solution [Tris-KCl buffer pH 9 ()] for anion exchange-based DNA extraction.](/image/article/2012/AY/c1ay05552f/c1ay05552f-f2.gif) | ||
Fig. 2 Graph showing the average νEOF at different applied voltages for the binding/wash solution [MES buffer pH 5 (■)] and elution solution [Tris-KCl buffer pH 9 ( )] for anion exchange-based DNA extraction. )] for anion exchange-based DNA extraction. | ||
Due to the anionic nature of the chitosan surface, bulk movement of solutions via EOF occurs towards the positive electrode. As the electrophoretic mobility of DNA in free solution is 3.75 × 10−4 cm2 V−1sec−1, this combines with EOF to generate efficient movement of DNA within the microfluidic device.16
DNA extraction capability
The efficiency of the DNA extraction process was evaluated as a function of the time over which the elution buffer was pumped over the solid-phase. DNA extraction efficiency was calculated as the amount of DNA recovered during the elution expressed as a percentage of the amount of DNA initially loaded onto the system. Maximum DNA yields were obtained when a 20 min elution was used, resulting in an average DNA extraction efficiency of 46.6% (±5.6%) when a potential of 100 V cm−1 was applied (Fig. 3).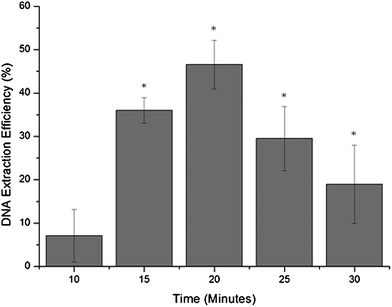 | ||
| Fig. 3 Graph showing DNA extraction efficiency as a function of the time the voltage was applied in order to perform the elution phase of the DNA extraction process on the microfluidic device, where * denotes eluted DNA samples which were successfully amplified by PCR (n = 3). | ||
The DNA extraction efficiency was also evaluated as a function of the voltage applied during the DNA elution phase using the optimised 20 min elution time (Fig. 4). The data showed a Gaussian shaped distribution, peaking at 100 V cm−1 with an average DNA extraction efficiency of 69.1% (±10.7%). The increase in average efficiency from Fig. 3 is due to the introduction of cooling the microfluidic device in order to minimize the effects of Joule heating.
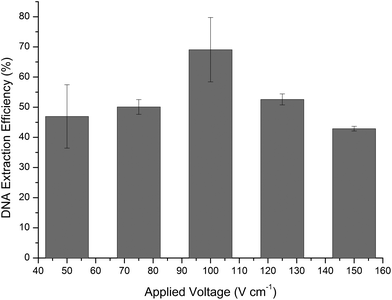 | ||
| Fig. 4 DNA extraction efficiencies expressed as a function of applied voltage for electrokinetic movement of reagents within the microfluidic system (n = 3). | ||
During the evaluation of DNA extraction efficiency, PCR reagents were loaded onto the microfluidic device within the DNA amplification chamber. Following the extraction process, the PCR reagents were removed and subject to DNA amplification on a conventional thermal cycler in order to assess whether the use of electrokinetic movement had any adverse affect on the reagents. An evaluation of these samples confirmed that the use of electrokinetic movement did not have an inhibitory effect on the PCR reagents. However, it was found that altering the voltages affected the relative PCR efficiency, as observed from a change in fluorescence intensity of the PCR products produced. The most efficient DNA amplification results were obtained when 100 V cm−1 was used, as this resulted in an average signal intensity at least 21% higher than at any other applied voltage.
Optimal conditions were achieved using an applied voltage of 100 V cm−1 for both DNA extraction and PCR efficiency. Whilst it is clear that the application of different voltages during the DNA extraction step has a direct effect on the efficiency of the process, there maybe direct and/or indirect influences on PCR efficiency. It is hypothesized that both play an important role, whereby the voltage can directly affect the PCR reagents and can also indirectly influence the efficiency of the upstream DNA extraction process resulting in differing quality and quantity of DNA for amplification.
Integrated DNA extraction and amplification
Following optimisation of the DNA extraction process and the confirmation that the PCR reagents were not inhibited by the electrokinetic movement, integration of DNA extraction and amplification on the microfluidic device was evaluated. Control of DNA movement between the DNA extraction and amplification chambers was driven electrokinetically and the eluted DNA was transferred directly into the PCR chamber. The PCR chamber was positioned on a thermoelectric Peltier which was used to provide the thermal cycling. The eluted DNA was successfully amplified on the integrated microfluidic system, as confirmed by off-chip capillary gel electrophoresis (Fig. 5).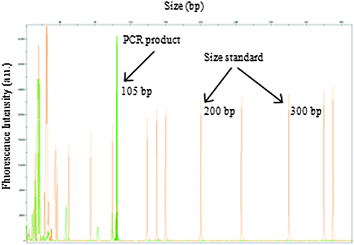 | ||
| Fig. 5 Electropherogram showing PCR products from the amplification of the Amelogenin locus (X = 105 bp) from a human urine sample, using DNA extracted and amplified on the integrated microfluidic device as confirmed on an ABI 3500 Genetic Analyzer. | ||
Conclusions
Exploitation of EOF with enhanced electrophoretic mobility to achieve electrokinetic pumping provided high DNA extraction efficiencies, using a chitosan-based solid-phase, along with successful DNA amplification. The DNA extraction efficiencies presented are comparable to those summarized in a recent review, which reported average efficiencies of between 60 and 70% for the majority of silica-bead and anion exchange-based microfluidic devices.3The ability to integrate both DNA extraction and amplification on a single device offers several advantages, including a reduction in the potential for contamination of the biological sample and reduced reagent consumption. While the DNA extraction efficiencies reported here are comparable to those previously described for electrokinetically driven systems, the integration with PCR was improved. By using an anion exchange matrix rather a than silica one, the solutions used are more compatible for integration with downstream processes as the use of chaotropic salts and organic solvents can be avoided.11 Moreover, the opportunity exists for further integration with downstream detection techniques such as DNA hybridisation or capillary electrophoresis.
By controlling the movement of reagents electrokinetically, moving components either on- or off-chip are eliminated which greatly reduces the complexity of the design and the footprint of the system. The possibility of incorporating reagent storage as previously described could also increase the application of the system described into ‘ready-to-use’ microfluidic devices.11
Acknowledgements
The authors would like to thank Dr Steve Clark for manufacturing the microfluidic devices, Dr Peter Docker for development of the Peltier thermal cycling system and Professor Nick Goddard for the kind gift of the carbon filled polystyrene electrodes.Notes and references
- G. M. Whitesides, Nature, 2006, 442, 368–373 CrossRef CAS.
- K. M. Horsman, J. M. Bienvenue, K. R. Blasier and J. P. Landers, J. Forensic Sci., 2007, 52, 784–799 CrossRef CAS.
- J. Wen, L. A. Legendre, J. M. Bienvenue and J. P. Landers, Anal. Chem., 2008, 80, 6472–6479 CrossRef CAS.
- K. A. Melzak, C. S. Sherwood, R. F. B. Turner and C. A. Haynes, J. Colloid Interface Sci., 1996, 181, 635–644 CrossRef CAS.
- S. M. Berry, E. T. Alarid and D. J. Beebe, Lab Chip, 2011, 11, 1747–1753 RSC.
- T. Nakagawa, T. Tanaka, D. Niwa, T. Osaka, H. Takeyama and T. Matsunaga, J. Biotechnol., 2005, 116, 105–111 CrossRef CAS.
- P. K. Dutta, J. Dutta and V. S. Tripathi, Journal of Scientific & Industrial Research, 2004, 63, 20–31 CAS.
- W. D. Cao, C. J. Easley, J. P. Ferrance and J. P. Landers, Anal. Chem., 2006, 78, 7222–7228 CrossRef CAS.
- C. R. Reedy, K. A. Hagan, B. C. Strachan, J. J. Higginson, J. M. Bienvenue, S. A. Greenspoon, J. P. Ferrance and J. P. Landers, Anal. Chem., 2010, 82, 5669–5678 CrossRef CAS.
- C. R. Reedy, C. W. Price, J. Sniegowski, J. P. Ferrance, M. Begley and J. P. Landers, Lab Chip, 2011, 11, 1603–1611 RSC.
- K. J. Shaw, D. A. Joyce, P. T. Docker, C. E. Dyer, G. M. Greenway, J. Greenman and S. J. Haswell, Lab on a Chip, 2011 Search PubMed.
- I. Erill, S. Campoy, N. Erill, J. Barbe and J. Aguilo, Sens. Actuators, B, 2003, 96, 685–692 CrossRef.
- H. Hartshorne, C. J. Backhouse and W. E. Lee, Sens. Actuators, B, 2004, 99, 592–600 CrossRef.
- T. Brooks and C. W. Keevil, Lett. Appl. Microbiol., 1997, 24, 203–206 CAS.
- B. E. Krenke, A. Tereba, S. J. Anderson, E. Buel, S. Culhane, C. J. Finis, C. S. Tomsey, J. M. Zachetti, A. Masibay, D. R. Rabbach, E. A. Amiott and C. J. Sprecher, Journal of Forensic Sciences, 2002, 47, 773–785 CAS.
- J. Chen, M. Wabuyele, H. Chen, D. Patterson, M. Hupert, H. Shadpour, D. Nikitopoulos and S. A. Soper, Anal. Chem., 2005, 77, 658–666 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2012 |