Screening for aromatase ligands in the extract of Broussonetia papyrifera using high performance liquid chromatography and ESI-mass spectrometry
Liping
Luo
a,
Liming
Shen
a,
Fang
Sun
a,
Yujie
Dai
b,
Heng
Zheng
c,
Zhongjun
Ma
*ad,
Ying
Xu
e and
Zengjun
Guo
*e
aSchool of Pharmaceutical Science, Zhejiang University, Zijingang Campus, No. 866 Yuhangtang Rd., Hangzhou, 310058, China
bKey Laboratory of Industrial Fermentation Microbiology (Tianjin University of Science & Technology), Ministry of Education, College of Bioengineering, Tianjin University of Science and Technology, Tianjin, 300457, P.R. China
cSchool of Life Science and Technology, China Pharmaceutical University, Nanjing, 210009, P.R. China
dInstitute of Marine Biology & Natural Products, Department of Ocean Science and Engineering Zhejiang University, Zijinggang Campus, No. 866 Yuhangtang Rd., Hangzhou, 310058, China. E-mail: mazj@zju.edu.cn; Fax: +86-571-88208540; Tel: +86-571-88208540
eDepartment of Pharmacy, School of Medicine, Xi'an Jiaotong University, 76 Yanta West Road, Xi'an, 710061, China
First published on 9th December 2011
Abstract
Aromatase (CYP19), which can catalyze the conversion of androgens to estrogens, has been one of the main inducers of hormone-related cancers. As a result, it is very important and necessary to find an aromatase inhibitor quickly and effectively. In this article, we screened for compounds that can bind to aromatase in the extract of Broussonetia papyrifera. The screening method HPLC/ESI-MS was employed in this presentation. Structures of twelve compounds were studied on the basis of LC-MS data. Six compounds were characterized as aromatase ligands through HPLC. Three of the ligands were identified as the glucosides of scopoletin, kaempferol and apigenin, whilst the others were vitexin, luteolin-7-O-β-D-glucopyranosid and dihydrochalcone. The bioassay indicated these compounds had aromatase inhibitory activity. We concluded that HPLC/ESI-MS was an effective means to reveal aromatase ligands. Moreover, five of the ligands found in this study were flavonoids. The C2, C3 double bond and the 7-OH were essential for the binding activity, which may be the binding sites for flavonoids.
Introduction
On a large scale, breast cancer is a major cause of death among women. A large portion of breast tumors are potentially endocrine-responsive, three quarters of cases occur in the post-menopausal period, while fewer are found in pre-menopausal women.1 Endocrine therapy, an effective approach for treating such tumors, has been used to decrease estrogen production. Aromatase (CYP19) is a key cytochrome P450 (CYP) enzyme, which catalyzes the rate-limiting aromatization step for the conversion of androgens to estrogens.2 The regulation and inhibition of aromatase activity have been seriously considered as drug targets because of the crucial role in regulating estrogen synthesis in post-menopausal women. In comparison with the estrogen antagonist tamoxifen,3,4aromatase inhibitors (AIs) have shown improved efficacy and reduced side effects against advanced and early stage breast cancer. Several classes of steroidal and non-steroidal AIs, such as aminoglutethimide and imidazole or triazole derivatives, have been designed in the past years.5–8 The first FDA-approved AI, aminoglutethimide, has shown some clinical benefits in breast cancer trials, however its lack of selectivity and weak aromatase inhibitory activity have limited its usefulness.6 Other synthetic compounds inhibit the synthesis of estrogen in tissues besides the ovaries also cause several severe adverse effects. In this context, natural products represent a promising source for the discovery of new drugs and templates for developing innovative AIs.9Broussonetia papyrifera (L.) L'Hér. ex Vent. (Moraceae) is a species of deciduous tree and its fruits have been used for to treat impotency and ophthalmic disorders.10,11 Extract of B. papyriera has shown antibacteria,12,13 antihepatotoxic,14antioxidant15 and lens aldose reductase inhibitory activities.11 Also, several flavonoid constituents of this plant have been shown to inhibit lipid peroxidation16 and to exhibit antiplatelet effects.17 Previous phytochemical work on this plant has resulted in the isolation of various types of flavonoids.18–20 Lee21 isolated five new active compounds with aromatase inhibition activity from the whole plants of B. papyrifera, using an in vitroaromatase inhibition assay.
In order to search for more compounds with aromatase inhibition activities, the structures of compounds from B. papyrifera were studied by LC-MS and screened for the aromatase ligands through HPLC.
Experimental
Materials and reagents
The leaves of Broussonetia papyrifera (Linn.) L'Hér. ex Vent. (Moraceae) were collected in Xi'an Oct 2006 and identified by Dr Z. J. Guo in Xi'an Jiaotong University. Naringenin was obtained from Best Biotech Inc. (Tianjin, China). All solvents used were HPLC grade, purchased from Merk Company (Germany). Aromatase was purchased from BD Biosciences (San Jose, USA). Ultrapure water in experiments was purified using a Milli-Q system (Millipore, Bedford, MA, USA).Instrumentation and analytical methods
The LC-MS analysis was carried out using a Finnigan LTQ linear ion trap mass spectrometer equipped with an ESI source. Full-scan mass spectra (m/z 120–2000) were collected in positive and negative ionization modes by ESI-MS. The column used was Zorbax SB-C18 (4.6 mm × 250 mm, 5 μm). The binary solvent gradient consisted of (A) water and (B) acetonitrile. The gradient program was as follows: initial A![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :B (95:5) lined from 0 min to 80 min to A:B (60:40), lined from 80 min to 110 min to A:B (20:80), lined from 110 min to 120 min to A:B (0:100). The flow rate was 0.5 mL min−1. The wavelength for detection was 203 nm by DAD detector with an injection volume of 20 μL. The HPLC analysis was conducted on an Agilent 1100, with the same column and chromatography conditions as LC-MS, elucidated above.
:B (95:5) lined from 0 min to 80 min to A:B (60:40), lined from 80 min to 110 min to A:B (20:80), lined from 110 min to 120 min to A:B (0:100). The flow rate was 0.5 mL min−1. The wavelength for detection was 203 nm by DAD detector with an injection volume of 20 μL. The HPLC analysis was conducted on an Agilent 1100, with the same column and chromatography conditions as LC-MS, elucidated above.
Extraction of B. papyrifera
The dried plant material (150 g) was ground and reflux extracted with 95% EtOH (2 × 4 L). The extracts were combined and concentrated under vacuum at 40 °C to yield 17.4 g dried extract.Preparation of HPLC-MS samples
Stock solution 95% ethanol extract of B. papyrifera (10.2 mg) was dissolved in 1 mL of MeOH and centrifuged at 10000 rpm for 10 min. The supernatant was collected as stock solution. The concentration was approximately 10 mg mL−1.
000 rpm, 10 min). The supernatant was collected for HPLC-MS analysis.
000 rpm, 30 min) and the lower liquid was collected for HPLC-MS analysis.
000 rpm, 30 min). The supernatant was collected for HPLC-MS analysis.
Results and discussion
Structures of compounds from the extract of B. papyrifera
Basing on the MS data in Table 1, twelve compounds were identified (see literature references in Table 1) from the extract of B. papyrifera, shown in Fig. 1. Their chromatograms and mass spectra are displayed in Fig. 2. Compounds identified in this article were almost all glycosides, a normal format of naturally occurring compounds, and their aglycones had been reported in previous studies. Flavonoids such as luteolin, apigenin, quercetin and so on had been isolated from B. papyrifera.13| No. | t R (min) | Formula | M. W. | MS data | Ref. |
|---|---|---|---|---|---|
| 1 | 32.15 | C21H18O12 | 462 | m/z 461([M − H]− 100%), 285(21), 923(62), 924(20); m/z 463([M + H]+ 100%), 287(21), 404(34), 420(47), 925(12) | 22 |
| 2 | 35.99 | C27H30O16 | 610 | m/z 609([M − H]− 100%), 447(48), 1219(17); m/z 611([M + H]+ 100%), 449(13), 1221(23) | 22 |
| 3 | 71.35 | C32H42O14 | 650 | m/z 649([M − H]− 100%), 325(10), 487(34) | 22 |
| 4 | 64.18 | C31H38O14 | 634 | m/z 633([M − H]− 100%), 309(17), 471(81) | 23 |
| 5 | 49.46 | C27H30O17 | 626 | m/z 625([M − H]− 100%), 301(12), 463(37) | 22 |
| 6 | 58.64 | C25H30O9 | 636 | m/z 635([M − H]− 100%), 311(8), 473(27), 1271(20) | 23 |
| 7 | 21.38 | C16H18O9 | 354 | m/z 353([M − H]− 100%), 191(15), 354(10), 707(12) | 24 |
| 8 | 24.35 | C21H20O12 | 464 | m/z 463([M − H]− 100%), 162(3), 285(9), 403(17) | 24 |
| 9 | 36.59 | C21H18O11 | 446 | m/z 445([M − H]− 100%), 269(24), 270(5), 891(92), 833(59), m/z 447([M + H]+ 100%), 271(21), 893(5) | 24 |
| 10 | 44.64 | C21H20O10 | 432 | m/z 431([M − H]− 100%), 311(2), 283(9), 863(17), m/z 433([M + H]+ 100%), 313(12) | 24 |
| 11 | 47.29 | C21H20O11 | 448 | m/z 447([M − H]− 100%), 285(6), 327(13), 895(65); m/z 449([M + H]+ 100%), 287(24), 376(29), 433(18), 897(6) | 24 |
| 12 | 100.61 | C20H22O5 | 342 | m/z 341([M − H]− 100%), 271(11), 342(18), 683(8) | 22 |
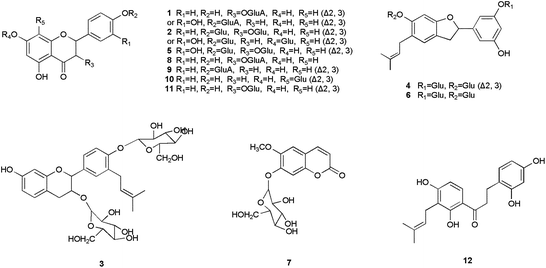 | ||
| Fig. 1 Structures of compounds from B. papyrifera. | ||
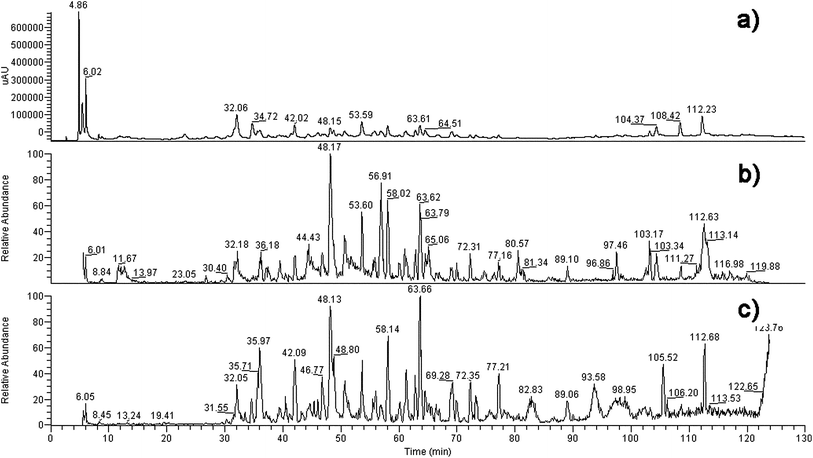 | ||
| Fig. 2 The chromatography (a), total ion current graph (−) (b) and total ion current graph (+) (c) of the compositions of the extract of B. papyrifera. | ||
The structural identification of the compounds were carried out on the basis of the MS data, referring to the reported compounds isolated from B. papyrifaer. Compound 1 was the glycoside of luteolin or kaempferol that had been isolated from B. papyrifaer. Compound 2 was the disaccharide of luteolin or kaempferol, while the aglycone of compound 5 was quercetin. The basic skeleton of compound 3 was broussinol.20 Compounds 4 and 6 had the same skeleton as moracin N.25 Compounds 7–9 were glucosides of scopoletin, kaempferol and apigenin, and compounds 10–11 were vitexin and luteolin 7-O-β-D-glucopyranoside, respectively, which had been isolated from the leaves of B. papyrifera before.26 Scopoletin and apigenin played as the aglycones of compounds 7 and 9. The aglycone of compound 8 was kaempferol.24 The glycosyls of compounds 8 and 9 were both glucuronic acid. Compounds 10 and 11 were identified as vitexin and luteolin 7-O-β-D-glucopyranoside with MS data consistent with literatures. Compound 12 owned the similar structure to 2,4,2′,4′-tetrahydroxy-3′-prenylchalcone.27 The only alternation of compound 12 lay in the conversion of an α,β-double bond to a single bond. Structures of 2,4,2′,4′-tetrahydroxy-3′-prenylchalcone, scopoletin, luteolin, kaempferol, apigenin and moracin N were shown in Fig. 3.
 | ||
| Fig. 3 Structures of compounds 2,4,2′,4′-tetrahydroxy-3′-prenylchalcone, scopoletin, luteolin, kaempferol, apigenin and moracin N. | ||
Positive experiment of naringenin
As the molecule weight was approximately 50 KDa, aromatase, together with the compounds that combined to aromatase, was cut off in ultra filtration centrifugation (molecular cut off: 10 KDa), which led to the reduced content of the compounds in the lower solution. Therefore, an HPLC-MS method was applied to screen aromatase ligands. In order to confirm whether the aromatase system was suitable for analysis or not, we selected naringenin28,29 as positive control for study. The peak area of naringenin in HPLC spectra was decreased by 40.2% after incubation with aromatase (Fig. 4). It was reported that naringenin could inhibit aromatase by 73.59 ± 0.18% at the concentration of 5 μM.29 As a consequence, it could be concluded that the system for screening aromatase ligands was reasonable.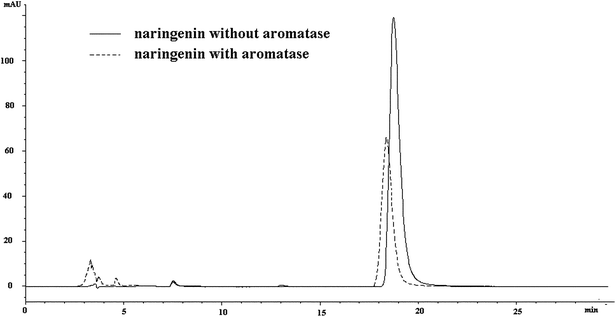 | ||
| Fig. 4 The chromatography of naringenin with and without incubation with aromatase. | ||
Aromatase ligands
An HPLC-MS method was applied to screen aromatase ligands. As the compounds that combined to aromatase were cut off in ultra filtration centrifugation (molecular cut off: 10 KDa), their contents in the lower solution would be reduced, so the peak areas of the compounds, which served as aromatase ligands, would decrease. According to the HPLC-UV spectrum (Fig. 5), compounds 1–6 did not bind to aromatase, while the other six compounds 7–12 did. Protein could not be retained in the reverse-phase column. The signals of the compounds that bond to aromatase would weaken or even disappear, presenting as the reduction of the peak areas at the same retention time. Thus it could be inferred the compounds whose areas reduced can bind to aromatase. According to the LC-MS results, reduction of areas happened at the retention times of 21.380, 24.356, 36.589, 44.642, 47.295, 100.610, 102.147, 110.009 min (Fig. 5). They were numbered as 7–12 in turn. The peak areas decreased by 71.06%, 27.11%, 90.57% and 53.98% for compounds 7, 8, 10 and 11, respectively, while the signals of 9 and 12 disappeared, indicating their binding capacities to aromatase. Beside which, the aglycones of compounds 8 and 11 (kaempferol and luteolin) were reported to inhibit aromatase.23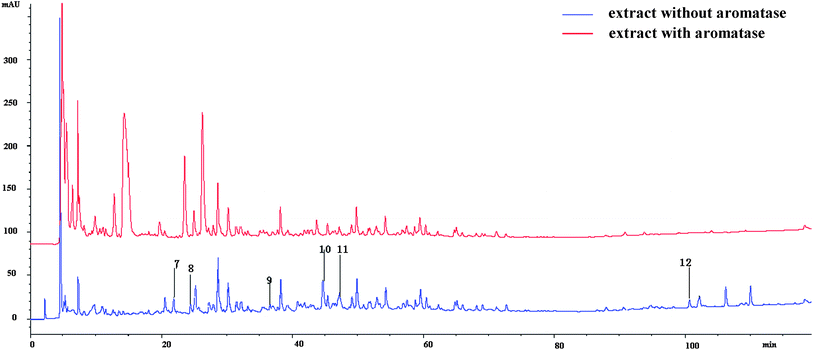 | ||
| Fig. 5 The chromatography of the compounds from B. papyrifera with or without incubation with aromatase. | ||
It was proved that there was no difference at the extract solubility between the two solvents, 10% MeOH and 100% MeOH. Due to the characteristic of aromatase as protein, it could be denatured if dissolved in 100% MeOH, so 10% MeOH was chosen as solvent instead of 100% MeOH to avoid this effect to aromatase. Furthermore, in order to investigate whether the signal of aromatase interrupted results, the HPLC analysis of aromatase was conducted separately and its signal was shown to have no impact on the results.
Five of the compounds binding to aromatase were identified as flavonoids. Flavonoids are a diverse group of phytochemicals produced in high quantities by various plants.30 Some flavonoids show estrogenic or anti-estrogenic activity, owing to the structural similarity with the estrogen skeleton. These flavonoids are able, like natural estrogens, to bind to the estrogen receptor and modulate its activity. They also block CYP19 (e.g., apigenin and kaempferol).24, 31, 32Flavonoids, having an anti-estrogenic effect, exhibited anti-cancer activity, especially in relation to tissues exposed to sex hormones (breast, prostate).32,33 Thus, flavonoids, as the steroidogenic enzyme inhibitors and the estrogen receptor modulators, were extensively studied for utilization in the prevention and treatment of some cancers. Lee21 isolated and identified fourteen compounds from B. papyrifera, all of which had aromatase inhibition activity and thirteen of which had flavonoid skeletons.
2,4,2′,4′-tetrahydroxy-3′-prenylchalcone 27 was shown to have aromatase inhibitory activity. Compared to 2,4,2′,4′-tetrahydroxy-3′-prenylchalcone, compound 12 without α,β-double bond also had strong binding capacity to aromatase and its signal in HPLC spectra disappeared completely (Fig. 5). Thus α,β-double bond of chalcones was not essential for binding with aromatase.24Flavonoids that bond to aromatase had 7-OH, relating to the ones did not bind to aromatase and demonstrating the significance of 7-OH in the binding activity – it might be the binding site for flavanoids. Flavanoids that bond to aromatase all possessed C2, C3 double bonds, demonstrating the significance of this double bond in binding to aromatase.
To further investigate the interaction between human aromatase and above coumarin and flavones, the molecular docking experiments were employed and the results were shown in Fig. 6. In the previous reports, it was suggested that porphyrin-Fe(III) and a series of amino acids in the active site cavity including Met374, Arg115, Ile133, Ala306, Thr310, Asp309, Val370 and Ser478, are responsible for important interactions between aromatase and its ligands.34 When it comes to compounds 7–12 in the present study, all the compounds could dock into the enzyme cavity specifically. The A ring of flavones and the benzene ring of coumarin contain π electron systems, which could form strong coordination bonds with heme ion. This situation was similar with most non steroidal aromatase inhibitors.35 Moreover, the hydroxy group in the B ring of flavones could form hydrogen bonds with amide back bone of Met 374 and/or Arg 115. Besides hydrogen bonds, the van der Waals interactions between compounds and some non-polar amino acids (e.g. Ile133, Ala306, Thr310) could further stabilize the binding of compounds with the enzyme active pocket. Therefore, the molecular docking experiments verified the results in the previous LC-MS analysis and screening.
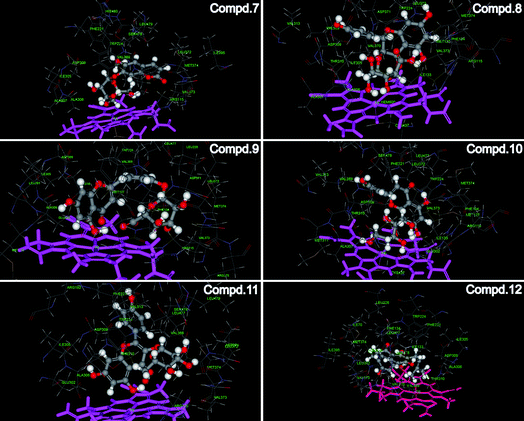 | ||
| Fig. 6 The interaction between human aromatase and compounds 7–12. Crystal structure of human aromatase was obtained from the RCSB protein data bank (http://www.pdb.org, pdb code: 3EQM). The molecular docking procedure was performed by using CDOCKER protocol for receptor-ligand interactions section of Discovery Studio 2.1. | ||
Conclusions
Broussonetia papyrifera, rich in flavonoids, has various bioactivities such as antifungal activity, antihepatotoxic activity, lens aldose reductase inhibition and so on. The aromatase inhibitors of it had been studied using an in vitroaromatase inhibitory assay. In this article, we applied HPLC-MS to screen aromatase ligands from B. papyrifera effectively and rapidly. Undoubtedly, it was a reasonable method of finding aromatase ligands from a complicated system such as the extract of traditional Chinese medicine without isolation. Six compounds were found to bind aromatase, five of which were flavonoids. This result showed that flavonoids had more affinity to aromatase. The structures of the flavonoids that could bind to the aromatase in this presentation indicated that the C2, C3 double bond and the 7-OH was essential for the binding activity, which suggests it might be the binding site for flavanoids. Furthermore, molecular docking experiments indicated that the π electron system of A ring and the hydroxy group of B ring were responsible for the reaction with aromatase. They formed strong coordination bonds and hydrogen bonds with the active site of aromatase, respectively, verifying the results in the previous LC-MS analysis and screening.Acknowledgements
The research was supported by National Scientific Foundation of China (NSFC No. 30701048) and Qianjiang Excellence Project of Zhejiang Province (2008R10035) and Zhejiang Key Science & Technological Program (2009C13028).References
- L. D. Maestro, M. Clavarezza and M. Venturini, Cancer Treat. Rev., 2007, 33, 681–687 Search PubMed.
- A. Maiti, M. Cuendet, V. L. Croy, D. C. Endringer, J. M. Pezzuto and M. Cushman, J. Med. Chem., 2007, 50, 2799–2806 Search PubMed.
- Z. W. Wong and M. J. Ellis, Br. J. Cancer, 2004, 90, 20–25 Search PubMed.
- A. Milla-Santos, L. Milla, J. Portella, L. Rollo, M. Pons, E. Rodeos, J. Casanovas and M. Puig-Gali, Am. J. Clin. Oncol., 2003, 26, 317–322 Search PubMed.
- J. Geisler and P. E. Lønning, Clin. Cancer Res., 2005, 11, 2809–2821 Search PubMed.
- L. B. Michaud, Am. J. Health Syst. Pharm., 2005, 62, 266–273 Search PubMed.
- R. W. Brueggemeier, J. C. Hackett and E. S. Diaz-Cruz, Endocr. Rev., 2005, 26, 331–345 Search PubMed.
- G. J. Kelloff, R. A. Lubet, R. Lieberman, K. Eisenhauer, V. E. Steele, J. A. Crowell, E. T. Hawk, C. W. Boone and C. C. Sigman, Cancer Epidemiol. Biomarkers Prev., 1998, 7, 65–78 Search PubMed.
- D. J. Newman, G. M. Cragg and K. M. Snader, J. Nat. Prod., 2003, 66, 1022–1037 CrossRef CAS.
- S. Chiang, Dictionary of Chinese Crude Drugs, Shanghai Scientific Technologic Publisher, Shanghai, 1986, pp. 2289 Search PubMed.
- H. Matsuda, H. Cai, M. Kubo, H. Tosa and M. Iinuma, Biol. Pharm. Bull., 1995, 18, 463–466 Search PubMed.
- S. Y. Kim, J. H. Kim, S. K. Kim, M. J. Oh and M. Y. Jung, J. Am. Oil Chem. Soc., 1994, 71, 633–640 Search PubMed.
- H. Y. Sohn, K. H. Son, C. S. Kwon, G. S. Kwon and S. S. Kang, Phytomedicine, 2004, 11, 666–672 CrossRef CAS.
- Y. S. Kim and K. H. Park, Kor. J. Pharmacogn., 1994, 25, 388–394 Search PubMed.
- A. Shirata and K. Takahashi, Sanshi Shikenjo Hokoku, 1982, 28, 691–705 Search PubMed.
- H. H. Ko, S. M. Yu, F. N. Ko, C. M. Teng and C. N. Lin, J. Nat. Prod., 1997, 60, 1008–1011 CrossRef CAS.
- C. N. Lin, C. M. Lu, H. C. Lin, S. C. Fang, B. J. Shieh, M. F. Hsu, J. P. Wang, F. N. Ko and C. M. Teng, J. Nat. Prod., 1996, 59, 834–838 Search PubMed.
- S. C. Fang, B. J. Shieh and C. N. Lin, Phytochemistry, 1994, 37, 851–853 Search PubMed.
- S. C. Fang, B. J. Shieh, R. R. Wu and C. N. Lin, Phytochemistry, 1995, 38, 535–537 Search PubMed.
- M. Takasugi, N. Niino, S. Nagao, M. Anetai, T. Masamune, A. Shirata and K. Takahashi, Chem. Lett., 1984, 13, 689 Search PubMed.
- D. Lee, K. P. Bhat, H. H. Fong, N. R. Farnsworth, J. M. Pezzuto and A. D. Kinghorn, J. Nat. Prod., 2001, 64, 1286–1293 CrossRef CAS.
- T. Hanato, H. Kagawa, T. Yasuhara and T. Okuda, Chem. Pharm. Bull., 1988, 36, 2090–2097 Search PubMed.
- C. Wang, T. Mäkelä, T. Hase, H. Adlercreutz and M. S. Kurzer, J. Steroid Biochem. Mol. Biol., 1994, 50, 205–212 CrossRef CAS.
- P. Hodek, P. Trefil and M. Stiborová, Chem.-Biol. Interact., 2002, 139, 1–21 Search PubMed.
- S. Matsuyama, Y. Kuwahara, S. Nakamura and T. Suzuki, Agric. Biol. Chem., 1991, 55, 1333–1341 CAS.
- X. H. Xu, S. H. Qian and M. G. Bian, Chin. J. Nat. Med., 2007, 5, 190–192 Search PubMed.
- G. Delle Monache, M. C. De Rosa, R. Scurria, A. Vitali, A. Cuteri, B. Monacelli, G. Pasqua and B. Botta, Phytochemistry, 1995, 39, 575–580 CrossRef.
- J. C. Le Bail, C. Pouget, C. Fagnere, J. P. Basly, A. J. Chulia and G. Habrioux, Life Sci., 2001, 68, 751–761 Search PubMed.
- F. Sun, L. Shen and Z. Ma, Food Chem., 2011, 126, 1337–1343 Search PubMed.
- R. A. Dixon and C. Steele, Trends Plant Sci., 1999, 4, 394–400 CrossRef.
- S. Chen, Front Biosci., 1998, 3, d922–933 Search PubMed.
- Y. S. Lee, B. J. Lorenzo, T. Koufis and M. M. Reidenberg, Clin. Pharmacol. Ther., 1996, 59, 62–67 Search PubMed.
- C. Nagata, N. Takatsuka, H. Shimizu, H. Hayashi, T. Akamatsu and K. Murase, Cancer Epidemiol. Biomarkers Prev., 2001, 10, 179–184 Search PubMed.
- P. R. Partha and R. Kunal, J. Mol. Model., 2010, 16, 1597–1616 Search PubMed.
- P. A. Cole and C. H. Robinson, J. Med. Chem., 1990, 33, 2933–2942 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2012 |