An organometallic approach for microporous organic network (MON)–Co3O4 composites: enhanced stability as anode materials for lithium ion batteries†
Han Sol
Lee
a,
Jaewon
Choi
a,
Jaewon
Jin
a,
Jiseul
Chun
a,
Sang Moon
Lee
b,
Hae Jin
Kim
b and
Seung Uk
Son
*a
aDepartment of Chemistry and Department of Energy Science, Sungkyunkwan University, Suwon 440-746, Korea. E-mail: sson@skku.edu; Fax: +82-031-290-4572; Tel: +82-031-299-5932
bKorea Basic Science Institute, Daejeon 350-333, Korea
First published on 7th November 2011
Abstract
Microporous organic network (MON)–Co3O4 composites were obtained via organometallic complexation of cobalt carbonyl with MONs and showed enhanced stability as anode materials due to the buffering effect of MONs.
Assembly between molecular building blocks is a powerful method for the preparation of diverse porous materials.1 For example, diverse metal–organic frameworks (MOFs) were prepared from organic building blocks and metal ions.2 However, the relative instability of MOFs against moisture or heat often limited their applications. Recently, for achieving the more stable porous materials, the pure organic networks were prepared using organic building blocks and organic linkers.3 For example, Cooper's group reported the formation of microporous organic materials by Sonogashira coupling between alkynes and aryl halides.4 The resultant permanent porosities of materials were controlled by changing the geometry of building blocks. In addition, the tailored functionalities can be incorporated into organic networks by designing the building blocks.5 For example, our group introduced the imidazolium salts into microporous organic networks and used them as heterogeneous catalysts in the carbon dioxide transformation into cyclic carbonates.5d Furthermore, considering the robust porosity of microporous organic networks, it can be expected that the secondary materials can be incorporated into these networks for diverse applications. However, as far as we are aware, this kind of approach was not reported.
Recently, energy storage has become a key issue in a broad range of research fields. As a very promising energy storage device, lithium ion batteries are attracting great attention of scientists.6 Extensive studies are actively underway for obtaining the more stable electrode materials and higher storage capacities. Metal oxides including Co3O4 have been continuously considered as one of the promising anode materials in lithium ion batteries.7 However, the insertion of lithium ions into Co3O4 led to a significant structural-distortion of the materials, which resulted in aggregation and ultimately a decrease of storage capacities.8 The existence of networks holding Co3O4 during lithium-insertion/desertion would be very helpful to maintain the storage capacities. In this work, we report the synthesis of microporous organic network (MON)–Co3O4 composites via an organometallic (well-established alkyne–cobalt carbonyl chemistry)9 approach for incorporating Co3O4 into organic networks and their applications as anode materials in lithium ion batteries.
Scheme 1 shows the overall synthetic route for MON–Co3O4 composites. First, 1,3-bis(4-iodophenyl)benzene was prepared from 1,3-diiodobenzene.10 Then, for the preparation of the targeted MONs, 1,3-bis(4-iodophenyl)benzene (0.12 g, 0.24 mmol) and tetrakis(4-ethynylphenyl)methane3e (50 mg, 0.12 mmol) were dissolved in a mixture of toluene (5 mL) and diisopropylamine (2 mL). After adding bis(triphenylphosphine)-palladium dichloride (10 mg, 0.014 mmol) and copper iodide (10 mg, 0.053 mmol), the reaction mixture was heated at 90 °C for 1 day. Then, the formed precipitates were retrieved by centrifugation and washed with methanol, methylene dichloride, toluene and hexane. The resultant powder was dried under vacuum.
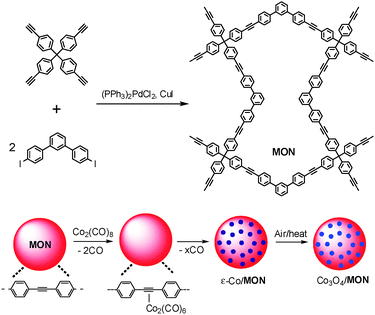 | ||
| Scheme 1 Synthesis of a microporous organic network (MON) and an organometallic route for preparation of MON–Co3O4 composites. | ||
Fig. 1a shows a typical scanning electron microscopy (SEM) image of the obtained MON. Overall, the materials had a spherical shape with 1.02 μm average size (Fig. 1a and b). Brunauer–Emmett–Teller (BET) analysis in Fig. 1c showed a 564 m2 g−1 surface area and micro-porosity (pore-size distribution based on a DFT method). Thermogravimetric analysis (TGA) showed that the materials start to decompose at 305 °C (Fig. 1d). Powder X-ray diffraction (PXRD) studies showed amorphous character of MONs, as observed in organic microporous materials in the literature3a (Fig. S2 in the ESI†). The chemical component of MON was further analyzed by solid-phase 13C-nuclear magnetic resonance (NMR) spectroscopy, which showed 13C peaks from the aromatic carbons at 150–120 ppm and the benzyl carbon at 65 ppm. Especially, as shown in Fig. 1e, the existence of internal alkynes was clearly proven by the 13C peak at 90 ppm.
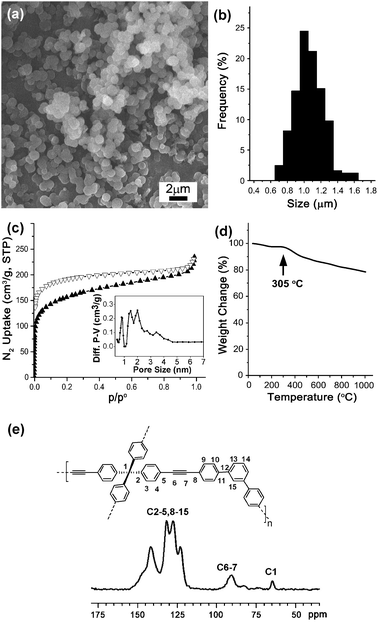 | ||
| Fig. 1 (a) SEM image; (b) size-distribution diagram; (c) BET analysis; (d) TGA curve; (e) solid-phase 13C-NMR spectrum of MON. | ||
Next, considering the porosity of MON and existence of alkyne groups, we tried to incorporate the cobalt species into the porous networks. It is well known that Co2(CO)8 can easily form stable alkyne–Co(CO)6 complexes by reacting with diverse alkynes including 1,2-diarylacetylene even at ambient temperature, which gradually lose carbonyl ligands at elevated temperature.9 Considering this, MON (30 mg) and Co2(CO)8 (30 mg, 0.035 mmol) were dispersed in toluene (5 mL). Then, the temperature was gradually increased to 90 °C. During heating, the yellow solid became slowly black. The reaction on solid was followed by infrared (IR) spectroscopy through extracting the solid materials from the reaction mixture during the reaction. After aging overnight, the reaction mixture was cooled to room temperature and the solid was retrieved by centrifugation.
Fig. 2a reveals the typical TEM image of the obtained black powder, which shows the formation of cobalt species on materials. Fig. 2b shows the IR studies νCO of isolated solids during the reaction. The new peaks at 2089, 2053 and 2027 cm−1 appeared gradually. These peaks were quite different from those (2068, 2041, 2024, 1992 and 1860 cm−1) of Co2(CO)8 and matched well with those (2089, 2055 and 2026 cm−1) of the 1,2-diphenylacetylene–Co2(CO)6 complex.9a The νCO peaks gradually disappeared to generate the metallic cobalt in the MON. The PXRD pattern of the black powder showed the very broad peaks at 44, 47 and 49° which are very close to those of epsilon Co11 (Fig. S2 in the ESI†). In the control experiment, when 700 nm-sized non-porous polystyrene12 beads were used instead of the MON, the cobalt species were not loaded homogeneously on the polymer particles, indicating the less efficient interaction between polymer particles and Co2(CO)8 possibly due to the absence of alkynes and porosity (Fig. S1 in the ESI†).
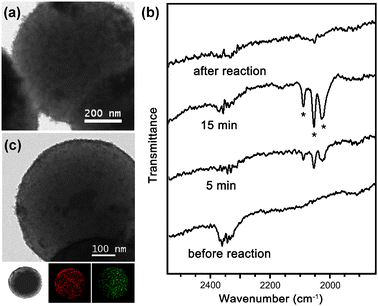 | ||
| Fig. 2 (a) TEM image of black powder (MON–Co); (b) IR studies of the reaction; (c) TEM/EDS-mapped images (red: cobalt, green: oxygen) of oxygen-treated black powder (MON–Co3O4, MC-30). | ||
To transform the metallic cobalt into cobalt oxides, the black powder was heated at 225 °C for 2 hours under oxygen and aged for 5 hours under argon. The formation of cobalt oxide species was characterized by PXRD studies. As shown in Fig. 3, the PXRD patterns matched well with that of Co3O4 (JCPDS #74-2120). Energy dispersive X-ray spectroscopy (EDS) with TEM studies showed the homogeneous distribution of cobalt and oxygen on black powder (Fig. 2c). BET analysis of composites showed the sharply-reduced surface area, compared with that of the MON (Fig. S3, ESI†). By increasing the precursor amount, 15 (MC-15), 23 (MC-23), 30 (MC-30) and 60 mg (MC-60) Co2(CO)8 per 30 mg MON, the Co3O4 contents in composites were systematically controlled (Fig. S1 in the ESI†). According to inductively coupled plasma-atomic emission spectroscopy (ICP-AES), the used precursors were quantitatively loaded on the MON. Due to the increased amount of cobalt species, the PXRD patterns became gradually sharper (Fig. 3).13
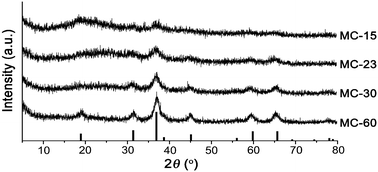 | ||
| Fig. 3 Powder X-ray diffraction patterns of MON–Co3O4 composites and Co3O4 in JCPDS #74-2021. | ||
To test the storage capacity of MON–Co3O4 composites as anode materials, the conventional coin cells were fabricated (see the ESI† for details). Then, the cells were discharged from an open-circuit voltage of 1 mV and cycled between 1 mV and 3.0 V at a current density of 50 mA g−1. Fig. 4 shows the cell performance of materials. (Also, see Fig. S3 in the ESI† for charge–discharge profiles and coulombic efficiencies.) It is noteworthy that the Co3O4 nanoparticles prepared by the same procedure without the MON (Fig. S1 in the ESI†) showed the sharp decrease of storage capacity (after 30 cycles, 90 mAh g−1) as observed in the literature.8 In comparison, the MON–Co3O4 composites showed the enhanced stability. Especially, after 30 cycles, MC-30 and MC-60 maintained the discharge capacity of 420 and 640 mAh g−1 respectively (cf. theoretical max. discharge capacity of graphite: 372 mAh g−1).14 Usually, the nano-sized Co3O4 materials were structurally distorted through lithium insertion and resulted in material breakdown and aggregation.8 However, the existence of the buffering networks in MON–Co3O4 composites would block the aggregation of materials, leading to the enhanced discharge stability.15 In addition, MON–Co3O4 composites showed the excellent coulombic efficiencies and rate capability, showing the stable performance at higher current densities (Fig. S4, ESI†).
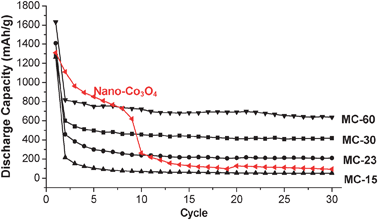 | ||
| Fig. 4 Cell cycle performance of MON–Co3O4 composites. | ||
In conclusion, microporous organic networks were prepared by Sonogashira coupling between tetrakis(4-ethynylphenyl)-methane and 1,3-bis(4-iodophenyl)benzene. MON–Co3O4 composites were prepared via organometallic complexation of cobalt carbonyl with MONs and the successive oxidation. MON–Co3O4 composites showed the enhanced stability as anode materials due to the buffering effect of MONs. Considering these observations, we believe that composites of MONs with diverse metal oxides are new candidates of anode materials for lithium ion batteries.
This work was supported by grant NRF-2009-0084799 through the National Research Foundation of Korea funded by the Ministry of Education, Science and Technology. JC thanks for grants NRF-2010-0029698 (Priority Research Centers Program) and R31-2008-10029 (WCU program). HJK thanks for KBSI grant P30402 and Hydrogen Energy R & D Center, a 21st century Frontier R & D Program.
Notes and references
- Review: J. R. Holst and A. I. Copper, Adv. Mater., 2010, 22, 5212 CrossRef CAS.
- Reviews: (a) C.-D. Wu and W. Lin, Angew. Chem., Int. Ed., 2007, 46, 1075 CrossRef CAS; (b) M. Dincá and J. R. Long, Angew. Chem., Int. Ed., 2008, 47, 6766 CrossRef.
- Review: (a) C. Weder, Angew. Chem., Int. Ed., 2008, 47, 448 CrossRef CAS. Selected recent examples: (b) A. Patra, J.-M. Koenen and U. Scherf, Chem. Commun., 2011, 47, 9612 RSC; (c) Z. Wang, B. Zhang, H. Yu, L. Sun, C. Jiao and W. Liu, Chem. Commun., 2010, 46, 7730 RSC; (d) J. Germain, F. Svec and J. M. J. Fréchet, Chem. Mater., 2008, 20, 7069 CrossRef CAS; (e) S. Yuan, S. Kirklin, B. Dorney, D.-J. B. Liu and L. Yu, Macromolecules, 2009, 42, 1554 CrossRef CAS; (f) N. B. McKeown, B. Ghanem, K. J. Msayib, P. M. Budd, C. E. Tattershall, K. Mahmood, S. Tan, D. Book, H. W. Langmi and A. Walton, Angew. Chem., Int. Ed., 2006, 45, 1804 CrossRef CAS.
- J.-X. Jiang, F. Su, A. Trewin, C. D. Wood, N. L. Campbell, H. Niu, C. Dickinson, A. Y. Ganin, M. J. Rosseinsky, Y. Z. Khimyak and A. I. Cooper, Angew. Chem., Int. Ed., 2007, 46, 8574 CrossRef CAS.
- (a) J. Weber and A. Thomas, J. Am. Chem. Soc., 2008, 130, 6334 CrossRef CAS; (b) E. L. Spitler and W. R. Dichtel, Nat. Chem., 2010, 2, 672 CrossRef CAS; (c) E. Klontzas, E. Tylianakis and G. E. Froudakis, J. Phys. Chem. C, 2009, 113, 21253 CrossRef CAS; (d) H. C. Cho, H. S. Lee, J. Chun, S. M. Lee, H. J. Kim and S. U. Son, Chem. Commun., 2011, 47, 917 RSC.
- Recent reviews on energy storage-materials: (a) F. Cheng, Z. Tao, J. Liang and J. Chen, Chem. Mater., 2008, 20, 667 CrossRef CAS; (b) P. G. Bruce, B. Scrosati and J.-M. Tarascon, Angew. Chem., Int. Ed., 2008, 47, 2930 CrossRef CAS.
- Examples of cobalt oxide anode materials: (a) B. Guo, C. Li and Z.-Y. Yuan, J. Phys. Chem. C, 2010, 114, 12805 CrossRef CAS; (b) X. Wang, L. Yu, X.-L. Wu, F. Yuan, Y.-G. Guo, Y. Ma and J. Yao, J. Phys. Chem. C, 2009, 113, 15553 CrossRef CAS; (c) Y. Li, B. Tan and Y. Wu, Nano Lett., 2008, 8, 265 CrossRef CAS.
- On the origin of capacity fading problem: J.-G. Kang, Y.-D. Ko, J.-G. Park and D.-W. Kim, Nanoscale Res. Lett., 2008, 3, 390 CrossRef CAS.
- (a) N. M. Boyle, A. C. Coleman, C. Long, K. L. Ronayne, W. R. Browne, B. L. Feringa and M. T. Pryce, Inorg. Chem., 2010, 49, 10214 CrossRef CAS; Recent examples of the Co–alkyne organometallic approach for functional materials: (b) E. C. Constable, O. Eich, D. Fenske, C. E. Housecroft and L. Johnston, Chem.–Eur. J., 2000, 6, 4364 CAS; (c) B. E. Hamaoui, L. Zhi, J. Wu, J. Li, N. T. Lucas, L. Z. Tomvic, U. Kolb and K. Mullen, Adv. Funct. Mater., 2007, 17, 1179 CrossRef.
- K. Kokado, Y. Tokoro and Y. Chujo, Macromolecules, 2009, 42, 9238 CrossRef CAS.
- V. F. Puntes, K. M. Krishnan and A. P. Alivisatos, Science, 2001, 291, 2115 CrossRef CAS.
- See the ESI† for preparation of cells.
- The analysis by Scherrer's equation for MC-30 and MC-60 revealed 4.0 and 4.8 nm sizes of cobalt oxides respectively.
- The MON without Co3O4 showed the discharge capacity of 98 mAh g−1 after 30 cycles (Fig. S5 in the ESI†).
- Examples for composite anode materials: (a) J. Choi, J. Jin, I. G. Jung, J. M. Kim, H. J. Kim and S. U. Son, Chem. Commun., 2011, 47, 5241 RSC; (b) D. Wang, D. Choi, J. Li, Z. Yang, Z. Nie, R. Kou, D. Hu, C. Wang, L. V. Saraf, J. Zhang, I. A. Aksay and J. Liu, ACS Nano, 2009, 3, 907 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, additional TEM images, PXRD patterns, charge–discharge profiles and rate performance. See DOI: 10.1039/c1cc15535k |
| This journal is © The Royal Society of Chemistry 2012 |