Phosphate ligands in the gold(I)-catalysed activation of enynes†
Mihai
Raducan
a,
María
Moreno
a,
Christophe
Bour
a and
Antonio M.
Echavarren
*ab
aInstitute of Chemical Research of Catalonia (ICIQ), Av. Països Catalans 16, 43007 Tarragona, Spain. E-mail: aechavarren@iciq.es
bDepartament de Química Analítica i Química Orgànica, Universitat Rovira i Virgili, C/Marcel.li Domingo s/n, 43007 Tarragona, Spain
First published on 4th November 2011
Abstract
Gold(I) forms neutral complexes with binol phosphates that are unreactive in the catalytic cyclisation of enynes. Reaction in protic solvents or activation by silver(I) restores the catalytic activity.
The reactive gold(I) species [AuL]+ in the catalytic activation of alkynes, allenes, and alkenes are often formed by chloride abstraction from [AuCl(L)] complexes using silver(I) salts.1 In 2007, Toste and coworkers demonstrated that achiral complexes [AuCl(L)] became enantioselective catalysts upon activation with silver binol phosphate salts in the cyclisation of allenols such as 1 to form tetrahydrofuran 2 (Scheme 1) and in related transformations of allenyl sulfonamides,2,3 as well as in the enantioselective synthesis of pyrazolidines, isoxazolidines and tetrahydrooxazines.4 It was postulated that an ion pair [Au(L)]+X− was formed in situ by reaction of [AuCl(L)] with the silver binol phosphate, although the resulting complexes were neither isolated nor characterized.5 The importance of ion pairing has been recognized in other contexts in gold catalysis.6 Complexes [(AuX)2(L–L)] and [Au2XCl(L–L)] (L–L = bidentate phosphine) were also active in the enantioselective cyclisation of hydroxy- and sulfonamidoallenes.7 Chiral phosphoric acids in combination with gold complexes have also been used in other enantioselective transformations.8,9
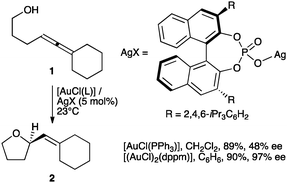 | ||
| Scheme 1 Enantioselective cyclisation of allenols.2 | ||
Despite the success in enantioselective catalysis using allene derivatives,2,4,7 this concept has not been extended to the activation of alkynes, the most general playground in gold catalysis.1 In addition, the nature of the silver(I) and gold(I) binol phosphate complexes used in the activation of allenes was not well defined. Here we show that the binol phosphates form covalent species [AuX(L)]. Although these complexes activate allenes, terminal alkynes lead to alkynyl–gold(I) complexes that are catalytically inactive.
Silver(I) phosphate complexes 5 and 6, prepared by reaction of the binol phosphoric acids 3 and 4 with Ag2O, showed 31P NMR spectra consistent with the formation of dimeric species or higher aggregates (Scheme 2).10 Thus, 5 shows an apparent triplet in CD2Cl2 or C6D6 at all concentrations. In diluted CD2Cl2 solutions, the 31P NMR spectrum of 6 consists of a triplet that becomes a multiplet and then a broad singlet in more concentrated solutions. The formation of dimeric species was confirmed by X-ray diffraction of complex 5, which showed a C2 symmetrical structure with a Ag–Ag distance of 3.072 Å (Scheme 2).
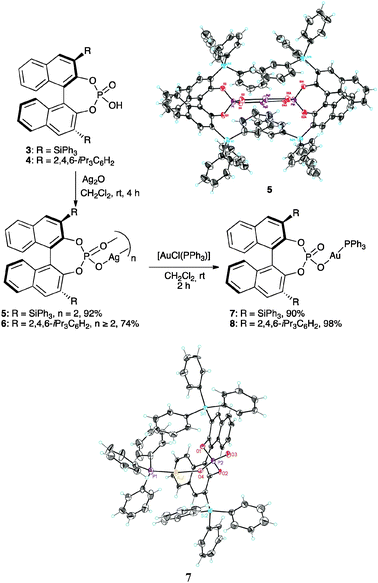 | ||
| Scheme 2 Synthesis and structures of Ag(I)–Au(I)–phosphate complexes. | ||
Silver(I) complexes 5 and 6 reacted with [AuCl(PPh3)] to give gold(I) complexes 7 and 8 in good yields (Scheme 2). These gold(I) phosphate complexes are very robust and can be purified by flash chromatography on SiO2. Long-range 31P–31P coupling could be observed in the 31P NMR spectra of 7 and 8 (3.6 and 2.6 Hz, respectively), which shows that the phosphate is covalently bound to Au(I). Their covalent structures were confirmed by the X-ray structure of complex 7·CHCl3, which shows an almost linear P–Au–O bond (172.7°). The Au–O distance of 2.056 Å is in line with reported data for other well-characterized phosphine–Au(I) phosphate complexes (2.06 Å).7a,11 The CHCl3 molecule is hydrogen bonded to the P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O group (ca. 1.93 Å). A second solvate containing water and methanol was obtained by crystallizing 7 from methanol in which the Au–O bond is lengthened from 2.056 Å in 7·CHCl3 to 2.101 Å in the 7·MeOH/H2O solvate. The PO distance in 7·MeOH/H2O is also longer than that in 7·CHCl3 (1.493 vs. 1.463 Å). Additionally, under concentrations comparable to the ones employed in catalysis, the molar conductivity of 7 in CH2Cl2 is two orders of magnitude lower than that of cationic gold(I) complex [Au(o-biphenPtBu2)(MeCN)]SbF6 (9a),12 which supports the covalent nature of 7 in solution.
O group (ca. 1.93 Å). A second solvate containing water and methanol was obtained by crystallizing 7 from methanol in which the Au–O bond is lengthened from 2.056 Å in 7·CHCl3 to 2.101 Å in the 7·MeOH/H2O solvate. The PO distance in 7·MeOH/H2O is also longer than that in 7·CHCl3 (1.493 vs. 1.463 Å). Additionally, under concentrations comparable to the ones employed in catalysis, the molar conductivity of 7 in CH2Cl2 is two orders of magnitude lower than that of cationic gold(I) complex [Au(o-biphenPtBu2)(MeCN)]SbF6 (9a),12 which supports the covalent nature of 7 in solution.
Well-characterized 7 and 8 (5 mol%) catalysed the cyclisation of allenol 1 into tetrahydrofuran 2 (80%, 3 h, 24% ee and 73% yield, 30 min, 48% ee, respectively) (Scheme 1) in total agreement with the results reported by Toste with the complexes formed in situ. Surprisingly, 7 and 8 were inactive in the cyclisation of 10, a highly reactive 1,6-enyne in gold(I)-catalyzed reactions.12,13 This enyne has been reported to form the product of single cleavage rearrangement 11 almost quantitatively with only 0.01 mol% of neutral complex [Au(NTf2)(PPh3)] in 30 min,14 whereas cationic catalyst [Au(o-biphenPCy2)(MeCN)]SbF6 led to 11 at temperatures as low as −63 °C12 (Scheme 3). In contrast, no reaction was observed after 3 days at 23 °C in the presence of complex 7. Similarly, no cyclisation of 13 into 1415 was observed even after heating at 40 °C in CH2Cl2 (microwave irradiation) with complexes 7 or 8 for 12 h. These reactions also failed with gold(I) phosphate complexes generated in situ by mixing Ag(I) complex 5 (2.5 mol%) with [AuCl(PPh3)] (5 mol%), [AuCl(o-biphenPCy2)] (5 mol%), [(AuCl)2(dppm)] (2.5 mol%), or [(AuCl)2(binap)] (2.5 mol%). Furthermore, no reaction was observed when enynol 15 was treated with 7 (5 mol%) in CH2Cl2 at 23–40 °C, whereas [Au(PPh3)Cl] and AgSbF6 (3 mol% each) catalysed the transformation of 15 into 1612 in 45 min (76% yield) (Scheme 3). The catalytic activity of 7 (5 mol%) was restored in the presence of MeOH (55 equivalents), leading to 16 (19%) and 17 (44%) after 48 h at 23 °C.
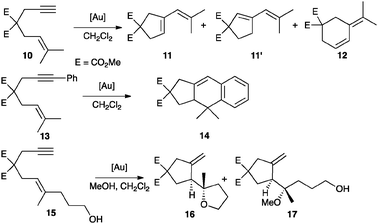 | ||
| Scheme 3 Gold(I)-catalysed cyclisations of selected 1,6-enynes. | ||
Addition of an equimolar amount of [Ag(NCMe)2]SbF616 to complex 7 (5 mol%) also restores the catalytic activity in the skeletal rearrangement of 1,6-enyne 10, yielding a 9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio of 11 and 12 after 20 min in quantitative yield at 23 °C. The formal intramolecular [4 + 2] cycloaddition of 13 also proceeded satisfactorily in the presence of [Ag(NCMe)2]SbF6 to give racemic 14 in 81% yield after 6 h. Presumably, under these conditions, catalytically active gold(I) complex [Au(PPh3)(MeCN)]SbF6 along with silver(I) salt 5 are formed.
:1 ratio of 11 and 12 after 20 min in quantitative yield at 23 °C. The formal intramolecular [4 + 2] cycloaddition of 13 also proceeded satisfactorily in the presence of [Ag(NCMe)2]SbF6 to give racemic 14 in 81% yield after 6 h. Presumably, under these conditions, catalytically active gold(I) complex [Au(PPh3)(MeCN)]SbF6 along with silver(I) salt 5 are formed.
Gold(I)–phosphate complexes 7 and 8 catalysed the hydration of 1-octyne in aqueous methanol at 23 °C (1 mol% catalysts, 67 h) leading to 2-octanone (43% and 100% yields with 7 and 8, respectively).17 Surprisingly, monitoring the reaction by 1H NMR revealed that deuterium exchange at the alkyne proton is much faster than the hydration reaction.
These results point to a facile deprotonation of the terminal alkyne by the gold(I) phosphate leading to the formation of the corresponding gold(I)–acetylide.18 Indeed, 1,6-enyne 10 reacted with 7 and 8 (5 mol%) in CH2Cl2 at 23 °C to form gold(I)–acetylide 18 and phosphoric acids 3 and 4 (43–47% conversion after 33 h). Phenylacetylene reacted similarly with complexes 7 and 8 to give PhC≡C–AuPPh3 (24–32% conversion after 17–25 h). Whereas acetylide 18, prepared from 10 and [Au(PPh3)Cl] in the presence of NaOEt, was stable in solution, the addition of 1 equivalent of TfOH or Tf2NH in CDCl3 led to the formation of 11 and 12, along with isomerized diene 11′19,20 (86%, 30 min and 80%, 90 min, respectively) (Scheme 4).
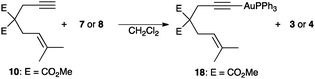 | ||
| Scheme 4 Formation of gold(I)–acetylide complex 18 from enyne 10. | ||
Neutral difluorophosphate complex [Au(OPOF2)(PPh3)] (19) has been recently prepared by reaction of [Au(PPh3)Cl] with AgPF6 in wet CH2Cl2.11,21Cyclisation of 1,6-enyne 10 with 19 as catalyst proceed very sluggishly under standard conditions (ca. 7% conversion after 1 h with 5 mol% 19 at 23 °C). This is somewhat surprising considering that the difluorophosphate anion is considerably less basic than the binol phosphates.22 For comparison, complexes [Au(PPh3)(MeCN)]SbF6 and [Au(NTf2)(PPh3)] (1 mol%) gave quantitatively 11, 11′, and 12 (2:1:2 ratio) in only 30 min. To further confirm the poor catalytic reactivity of covalently bound gold(I) phosphate complexes, we prepared cationic and neutral gold(I) complexes 9b and 9c (Fig. 1) and compared their reactivity in the cyclisation of 10. Thus, whereas 9b (5 mol%) led to quantitative conversions after 30 min, reaction with 9c required 17 h to give a different mixture of products.23,24
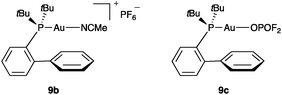 | ||
| Fig. 1 Cationic and neutral gold(I)–phosphate complexes 9b–c. | ||
Gold(I) complexes [AuX(L)] with phosphates and other anionic X ligands that are sufficiently basic form alkynyl–gold(I) complexes II and/or binuclear derivatives resulting from coordination of AuL+ to II,18a,d,f which are catalytic dead ends (Scheme 5). In contrast, these species probably play a minor role in catalysis with complexes [AuX(L)] or [Au(L)(L′)]X whose anionic ligands are the conjugate bases of very strong acids, such as HSbF6, HBF4, or Tf2NH. In these cases, the released strong Brønsted acid HX shifts the equilibrium towards I and does not act as a catalyst in the cyclisation as demonstrated by control experiments carried out with HBF4, TfOH, and Tf2NH and enynes 10 and 13.25
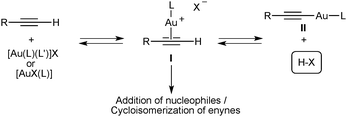 | ||
| Scheme 5 General pathways in the gold(I)-activation of terminal alkynes. | ||
In the case of gold(I)–phosphate complexes the use of a protic solvent such as methanol restores the catalytic activity presumably by facilitating the associative ligand substitution step12 through activation of the phosphate ligand by an H-bond and by lowering the basicity of the phosphate anions by solvation. The lack of reactivity of enyne 13 in the presence of 7 and 8 shows that the first step of the catalytic cycle that forms cationic species I is much slower with these neutral complexes than with cationic gold(I) catalysts [Au(L)(L′)]+X− (L′ = weakly coordinating ligand).
This work shows that in order to extend the chiral counterion concept to gold(I)-catalysed activation of alkynes, anionic ligands less basic than phosphates should be used.
We thank the MICINN (CTQ2010-16088/BQU and Consolider Ingenio 2010, Grant CSD2006-0003), the AGAUR (2009SGR47), Dr E. Escudero-Adán (ICIQ X-Ray Diffraction unit), and the ICIQ Foundation for financial support.
Notes and references
- (a) E. Jiménez-Núñez and A. M. Echavarren, Chem. Rev., 2008, 108, 3326–3350 CrossRef; (b) D. J. Gorin, B. D. Sherry and F. D. Toste, Chem. Rev., 2008, 108, 3351–3378 CrossRef CAS; (c) A. Fürstner, Chem. Soc. Rev., 2009, 38, 3208–3221 RSC.
- G. L. Hamilton, E. J. Kang, M. Mba and F. D. Toste, Science, 2007, 317, 496–499 CrossRef CAS.
- Enantioselective cyclisation of allenyl sulfonamides with (R)-xylyl-BINAP(AuOPNB)2 (PNB = p-nitrobenzoate): (a) R. L. LaLonde, B. D. Sherry, E. J. Kang and F. D. Toste, J. Am. Chem. Soc., 2007, 129, 2452–2453 CrossRef CAS; (b) J. H. Kim, S.-W. Park, S. R. Park, S. Lee and E. J. Kang, Chem.–Asian J., 2011, 6, 1982–1986 CrossRef CAS.
- R. L. La Londe, Z. J. Wang, M. Mba, A. D. Lackner and F. D. Toste, Angew. Chem., Int. Ed., 2010, 49, 598–601 CAS.
- Review on the use of chiral anions in stereoselective synthesis: J. Lacour and D. Moraleda, Chem. Commun., 2009, 7073–7089 RSC.
- D. Zuccaccia, L. Belpassi, F. Tarantelli and A. Macchioni, J. Am. Chem. Soc., 2009, 131, 3170–3171 CrossRef CAS.
- (a) K. Aikawa, M. Kojima and K. Mikami, Angew. Chem., Int. Ed., 2009, 48, 6073–6077 CrossRef CAS; (b) K. Aikawa, M. Kojima and K. Mikami, Adv. Synth. Catal., 2010, 352, 3131–3135 CrossRef CAS.
- (a) X.-Y. Liu and C.-M. Che, Org. Lett., 2009, 11, 4204–4207 CrossRef CAS; (b) Z.-Y. Han, H. Xiao, X.-H. Chen and L.-Z. Gong, J. Am. Chem. Soc., 2009, 131, 9182–9183 CrossRef CAS; (c) M. E. Muratore, C. A. Holloway, A. W. Pilling, R. I. Storer, G. Trevitt and D. J. Dixon, J. Am. Chem. Soc., 2009, 131, 10796–10797 CrossRef CAS; (d) C. Wang, Z.-Y. Han, H.-W. Luo and L.-Z. Gong, Org. Lett., 2010, 12, 2266–2269 CrossRef CAS; (e) A. S. K. Hashmi and C. Hubbert, Angew. Chem., Int. Ed., 2010, 49, 1010–1012 CrossRef CAS.
- Review on binol-derived phosphoric acids and amides as Brønsted acid catalysts: M. Rueping, B. J. Nachtsheim, W. Ieawsuwan and I. Atodiresei, Angew. Chem., Int. Ed., 2011, 50, 6706–6720 CrossRef CAS.
- X-Ray structure of a related complex: M. Rueping, R. M. Koenigs and I. Atodiresei, Chem.–Eur. J., 2010, 16, 9350–9365 CrossRef CAS.
- S. M. Kim, J. H. Park and Y. K. Chung, Chem. Commun., 2011, 47, 6719–6721 RSC.
- C. Nieto-Oberhuber, S. López, M. P. Muñoz, D. J. Cárdenas, E. Buñuel, C. Nevado and A. M. Echavarren, Angew. Chem., Int. Ed., 2005, 44, 6146–6148 CrossRef CAS.
- C. Nieto-Oberhuber, M. P. Muñoz, S. López, E. Jiménez-Núñez, C. Nevado, E. Herrero-Gómez, M. Raducan and A. M. Echavarren, Chem.–Eur. J., 2006, 12, 1677–1693 CrossRef CAS.
- N. Mézailles, L. Ricard and F. Gagosz, Org. Lett., 2005, 7, 4133–4136 CrossRef.
- (a) C. Nieto-Oberhuber, S. López and A. M. Echavarren, J. Am. Chem. Soc., 2005, 127, 6178–6179 CrossRef CAS; (b) C. Nieto-Oberhuber, P. Pérez-Galán, E. Herrero-Gómez, T. Lauterbach, C. Rodríguez, S. López, C. Bour, A. Rosellón, D. J. Cárdenas and A. M. Echavarren, J. Am. Chem. Soc., 2008, 130, 269–279 CrossRef CAS.
- M. Raducan, C. Rodríguez-Escrich, X. C. Cambeiro, E. Escudero-Adán, M. A. Pericàs and A. M. Echavarren, Chem. Commun., 2011, 47, 4893–4895 RSC.
- (a) N. Marion, R. S. Ramón and S. P. Nolan, J. Am. Chem. Soc., 2009, 131, 448–449 CrossRef CAS; (b) A. Leyva and A. Corma, J. Org. Chem., 2009, 74, 2067–2074 CrossRef CAS.
- (a) P. H.-Y. Cheong, P. Morganelli, M. R. Luzung, K. N. Houk and F. D. Toste, J. Am. Chem. Soc., 2008, 130, 4517–4526 CrossRef CAS; (b) Y. Odabachian, X.-F. Le Goff and F. Gagosz, Chem.–Eur. J., 2009, 15, 8966–8970 CrossRef CAS; (c) S. Gaillard, A. M. Z. Slawin and S. P. Nolan, Chem. Commun., 2010, 46, 2742–2744 RSC; (d) T. N. Hooper, M. Green and C. A. Russell, Chem. Commun., 2010, 46, 2313–2315 RSC; (e) G. C. Fortman, A. Poater, J. W. Levell, S. Gaillard, A. M. Z. Slawin, I. D. W. Samuel, L. Cavallo and S. P. Nolan, Dalton Trans., 2010, 39, 10382–10390 RSC; (f) A. Simonneau, F. Jaroschik, D. Lesage, M. Karanik, R. Guillot, M. Malacria, J.-C. Tabet, J.-P. Goddard, L. Fensterbank, V. Gandon and Y. Gimbert, Chem. Sci., 2011 10.1039/c1sc00478f; (g) M. C. Blanco, J. Cámara, M. C. Gimeno, P. G. Jones, A. Laguna, J. M. López de Luzuriaga, M. E. Olmos and M. D. Villacampa, Organometallics, 2011 DOI:10.1021/om200397t; (h) T. J. Brown and R. A. Widenhoefer, Organometallics, 2011 DOI:10.1021/om200840g.
- C. Nieto-Oberhuber, M. Paz Muñoz, S. López, E. Jiménez-Núñez, C. Nevado, E. Herrero-Gómez, M. Raducan and A. M. Echavarren, Chem.–Eur. J., 2006, 12, 1677–1693 CrossRef CAS.
- In addition, products assigned to the addition of TfOH or Tf2NH to the 1,6-enyne were also observed in the crude reaction mixtures.
- Complex 19 shows two signals in the 31P NMR (CDCl3) at δ 30.54 (s, PPh3) and −10.28 (t, J(31P–19F) = 973.0 Hz, PO2F2).
- (a) A pKa of 0.3 has been reported for difluorophosphoric acid in water: J. W. Larson and B. Su, J. Chem. Eng. Data, 1994, 39, 33–35 CrossRef CAS; (b) A pKa of 4.2 has been determined for phosphoric acid 6 in DMSO: P. Christ, A. G. Lindsay, S. S. Vormittag, J.-M. Neudörf, A. Berkessel and A. C. O'Donoghoue, Chem.–Eur. J., 2011, 17, 8524–8528 CrossRef CAS.
- See ESI† for details.
- (a) A pKa of 4.2 has been determined for phosphoric acid 6 in DMSO: P. Christ, A. G. Lindsay, S. S. Vormittag, J.-M. Neudörf, A. Berkessel and A. C. O'Donoghoue, Chem.–Eur. J., 2011, 17, 8524–8528 CrossRef CAS; (b) The pKa of benzoic acid in DMSO is 11: W. N. Olmstead and F. D. Bordwell, J. Org. Chem., 1980, 45, 3299–3305 CrossRef CAS.
- No reaction was observed with 10 or 13 using HBF4, TfOH, and Tf2NH (1 and 5 mol%, respectively, 24 h in CDCl3).
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis of starting substrates, characterization and X-ray crystallographic data. CCDC 841519 (5), 841517 (7·CHCl3), and 841518 (7·MeOH/H2O). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c1cc15739f |
| This journal is © The Royal Society of Chemistry 2012 |