Selective binding and fluorescence sensing of diphosphate in H2OviaZn2+-induced allosteric regulation of the receptor structure†
Carla
Bazzicalupi
a,
Andrea
Bencini
*a,
Stefano
Puccioni
a,
Barbara
Valtancoli
a,
Paola
Gratteri
b,
Alessandra
Garau
c and
Vito
Lippolis
*c
aDipartimento di Chimica “Ugo Schiff”, Università di Firenze, Via della Lastruccia 3, 50019-Sesto Fiorentino, Firenze, Italy. E-mail: andrea.bencini@unifi.it; Fax: +39 055 5573264; Tel: +39 055 3573264
bDipartimento di Scienze Farmaceutiche, Università di Firenze, Via Ugo Schiff, 50019-Sesto Fiorentino, Firenze, Italy
cDipartimento di Chimica Inorganica ed Analitica, Università degli Studi di Cagliari, S.S. 554 bivio per Sestu, 09042 Monserrato (CA), Italy. E-mail: lippolis@unica.it; Tel: +39 070 675 4467
First published on 7th November 2011
Abstract
A terpyridine-based receptor featuring two [9]aneN3 units is able to selectively bind and sense diphosphate over mono- and triphosphate in aqueous solution at pH 7, thanks to the conformational change of its structure induced by Zn2+ coordination to the polypyridyl moiety.
Selective binding and sensing of inorganic phosphate anions with synthetic receptors in aqueous solutions is one of the major targets of supramolecular chemistry,1 due to relevance of these anions in both biological2 and environmental chemistry.3 The design of metal-based receptors containing a fluorogenic unit represents one of the most common approaches to develop selective anion chemosensors.1 Selective Zn2+-based fluorescent chemosensors for inorganic phosphates in aqueous solution have been designed,1,4 exploiting the ability of this metal to form coordinative bonds with these anions, the fluorescence emission being generally controlled by the direct binding of the anionic species to the metal. However, in principle metal cations can also be used to organize the receptor structure to improve its binding ability and/or to impart selectivity in anion recognition through weak forces, without a direct participation of the metal cation in the binding process. Although this structural role of the metal cation is common in transport proteins or enzymes,5 its use in anion coordination chemistry is rare;6 in particular, to the best of our knowledge, a Zn2+ allosteric role has not yet been exploited for simultaneous recognition and sensing of anionic species, such as phosphates, by synthetic receptors in H2O.
We have recently reported that receptors composed of two [9]aneN3 units linked by rigid heteroaromatic units (1–3 in Scheme 1) are able, when protonated, not only to form remarkably stable 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 adducts with phosphate anions (monophosphate (MP), diphosphate (DP) and triphosphate (TP)) in H2O, but also to selectively recognize these anions on a size-base following a host–guest geometrical complementarity.7 In fact, the anions are encapsulated within the cleft delimited by the two protonated [9]aneN3 units, whose dimension is determined by the length of the heteroaromatic spacer. In consequence, receptor 1, which possesses the larger cavity, selectively binds TP over MP and DP, while 3 displays selectivity for the smaller MP. Finally, 2 shows a slight preference for DP binding, due to its middle-sized cavity. With this in mind, we have now synthesized receptor L (see ESI†), in which two [9]aneN3 moieties are bridged by a terpyridine (tpy) unit. Similarly to 1–3, L easily protonates in aqueous solution affording polyammonium cations (at neutral pH the receptor is present in solution mainly in its diprotonated form, (H2L)2+, see ESI†), a potential host species for anions. Despite these characteristics, potentiometric titrations show that L possesses a very weak binding ability for inorganic phosphates, the formation constants of the 1:1 adducts with both DP and TP being far lower than those of the corresponding complexes with 1–3, while MP complexation is not detected in H2O (see ESI†). Overall, the receptor does not display any ability to discriminate between DP and TP (log K = 2.9(1) and 2.7(1) for the equilibria (H2L)2+ + P2O74− = [H2L(P2O7)]2− and (H2L)2+ + P3O105− = [H2L(P3O10)]3−, respectively). At the same time, 1H NMR measurements carried out at pH 7 in D2O show that the signals of L are almost unaffected by the presence of MP, DP or TP: a small downfield shift, lower than 0.05 ppm, is observed only in the presence a 10-fold excess of DP or TP. This scarce binding ability can be due to the possible different conformations that L can assume in solution (Scheme 1). In fact, the trans–trans conformation (the most stable in aqueous solution for tpy)8 brings the two [9]aneN3 moieties to set far from each other, thus preventing the formation of clefts/pockets of suitable dimension for an efficient substrate encapsulation. This hypothesis is confirmed by the UV-Vis spectrum of L at neutral pH, which shows the typical band of the trans–transtpy conformation at ca. 295 nm. Under these conditions the system is also non-emissive. These spectral features are not affected by addition of MP, DP or TP, even in large excess (50 eq.). Conversely, addition of increasing amounts of Zn2+ to a solution of L at pH 7 (TRIS buffer), up to a 1:1 Zn2+/L molar ratio, causes the progressive disappearance of the absorption band at 295 nm and the formation of a new band at 315 nm, attributable to the conversion of the ligand from the trans–trans to the cis–cis conformation, induced by Zn2+ binding by the tpy unit of L (Fig. 1a). Potentiometric titrations detect the formation of a stable mononuclear [ZnL]2+ complex in aqueous solution (log K = 16.81 for the equilibrium Zn2+ + L = [ZnL]2+). Interestingly, this complex shows a marked tendency to protonate in aqueous solution and, actually, the diprotonated complex [ZnLH2]4+ is the only species present in solution at neutral pH (see ESI†). In this complex, the acidic protons are likely to be located on the [9]aneN3 units. Ligand L can also form, in the presence of an excess of Zn2+, di- and trinuclear complexes. However, the equilibrium constants for the addition of a second and a third Zn2+ cation to [ZnL]2+ are far lower than the formation constant of the 1:1 complex and polynuclear species are not formed in the presence of 1 eq. of Zn2+ (see ESI†).
:1 adducts with phosphate anions (monophosphate (MP), diphosphate (DP) and triphosphate (TP)) in H2O, but also to selectively recognize these anions on a size-base following a host–guest geometrical complementarity.7 In fact, the anions are encapsulated within the cleft delimited by the two protonated [9]aneN3 units, whose dimension is determined by the length of the heteroaromatic spacer. In consequence, receptor 1, which possesses the larger cavity, selectively binds TP over MP and DP, while 3 displays selectivity for the smaller MP. Finally, 2 shows a slight preference for DP binding, due to its middle-sized cavity. With this in mind, we have now synthesized receptor L (see ESI†), in which two [9]aneN3 moieties are bridged by a terpyridine (tpy) unit. Similarly to 1–3, L easily protonates in aqueous solution affording polyammonium cations (at neutral pH the receptor is present in solution mainly in its diprotonated form, (H2L)2+, see ESI†), a potential host species for anions. Despite these characteristics, potentiometric titrations show that L possesses a very weak binding ability for inorganic phosphates, the formation constants of the 1:1 adducts with both DP and TP being far lower than those of the corresponding complexes with 1–3, while MP complexation is not detected in H2O (see ESI†). Overall, the receptor does not display any ability to discriminate between DP and TP (log K = 2.9(1) and 2.7(1) for the equilibria (H2L)2+ + P2O74− = [H2L(P2O7)]2− and (H2L)2+ + P3O105− = [H2L(P3O10)]3−, respectively). At the same time, 1H NMR measurements carried out at pH 7 in D2O show that the signals of L are almost unaffected by the presence of MP, DP or TP: a small downfield shift, lower than 0.05 ppm, is observed only in the presence a 10-fold excess of DP or TP. This scarce binding ability can be due to the possible different conformations that L can assume in solution (Scheme 1). In fact, the trans–trans conformation (the most stable in aqueous solution for tpy)8 brings the two [9]aneN3 moieties to set far from each other, thus preventing the formation of clefts/pockets of suitable dimension for an efficient substrate encapsulation. This hypothesis is confirmed by the UV-Vis spectrum of L at neutral pH, which shows the typical band of the trans–transtpy conformation at ca. 295 nm. Under these conditions the system is also non-emissive. These spectral features are not affected by addition of MP, DP or TP, even in large excess (50 eq.). Conversely, addition of increasing amounts of Zn2+ to a solution of L at pH 7 (TRIS buffer), up to a 1:1 Zn2+/L molar ratio, causes the progressive disappearance of the absorption band at 295 nm and the formation of a new band at 315 nm, attributable to the conversion of the ligand from the trans–trans to the cis–cis conformation, induced by Zn2+ binding by the tpy unit of L (Fig. 1a). Potentiometric titrations detect the formation of a stable mononuclear [ZnL]2+ complex in aqueous solution (log K = 16.81 for the equilibrium Zn2+ + L = [ZnL]2+). Interestingly, this complex shows a marked tendency to protonate in aqueous solution and, actually, the diprotonated complex [ZnLH2]4+ is the only species present in solution at neutral pH (see ESI†). In this complex, the acidic protons are likely to be located on the [9]aneN3 units. Ligand L can also form, in the presence of an excess of Zn2+, di- and trinuclear complexes. However, the equilibrium constants for the addition of a second and a third Zn2+ cation to [ZnL]2+ are far lower than the formation constant of the 1:1 complex and polynuclear species are not formed in the presence of 1 eq. of Zn2+ (see ESI†).
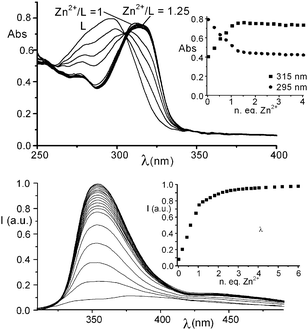 | ||
| Fig. 1 (a) UV-Vis spectra and absorbances at 315 and 295 nm of L (inset) in H2O in the presence of increasing amounts of Zn2+ (L = 1.0 × 10−4 M); (b) fluorescence emission spectra and emission intensity at 355 nm (inset) of L in H2O in the presence of increasing amounts of Zn2+ (L = 1.0 × 10−5 M). | ||
As a matter of fact, a mononuclear [ZnLH2](ClO4)4·2H2O complex (I) can be isolated as a solid compound from solutions containing L and Zn2+ in equimolecular ratio. More interestingly, Zn2+ binding strongly affects the fluorescence emission of L. In fact, addition of increasing amounts of Zn2+ leads to a marked enhancement of the fluorescence emission of the tpy unit of L up to a 1:1 Zn2+/L molar ratio (Fig. 1b). A further slight increase of the emission is also observed at higher Zn2+/L molar ratios, probably due to the formation of di- or trinuclear complexes.
Potentiometric measurements point out that, differently from L, its mononuclear Zn2+ complex shows a marked tendency to bind DP, the equilibrium constants for the addition of DP to the 1:1 Zn2+ complex of L being remarkably higher than those for the addition of DP to L in the absence of Zn2+. In particular, the protonated species [ZnLH2]4+ forms a remarkably stable [ZnLH2(P2O7)] neutral adduct (log K = 6.95 for the equilibrium [ZnLH2]4+ + P2O74− = [ZnLH2(P2O7)], see ESI†). The binding ability of the same Zn2+ complex for MP and TP is remarkably lower. This behavior can be better visualized by considering a competitive system containing the 1:1 Zn2+ complex of L and the three anions in equimolecular concentrations and calculating the overall percentages of the different adducts over a wide pH range.9 The 1:1 Zn2+ complex of L selectively binds DP over MP and TP in a wide pH range (Fig. 2). For instance at pH 7, the percentage of complexed DP is ca. 90%, while the percentages of MP and TP bound to the Zn2+ complex are ca. 2% and 8% respectively.
![Plots of the overall percentages of MP, PP and TP complexed by the 1 : 1 Zn2+ complex of L in H2Ovs. pH ([L] = [Zn2+] = [PO43−] = [P2O74−] = [P3O105−] = 1.0 × 10−3 M).](/image/article/2012/CC/c1cc15934h/c1cc15934h-f2.gif) | ||
| Fig. 2 Plots of the overall percentages of MP, PP and TP complexed by the 1:1 Zn2+ complex of L in H2Ovs. pH ([L] = [Zn2+] = [PO43−] = [P2O74−] = [P3O105−] = 1.0 × 10−3 M). | ||
This recognition ability for DP can be reasonably ascribed to an optimal dimensional fitting of this anion within the cleft between the two [9]aneN3 units, generated by the ligand conformational change upon Zn2+ binding. This suggestion is supported by 1H NMR spectra recorded on D2O solutions of I in the presence of the different anions at pH 7, which shows a 0.22, 0.23 and 0.07 ppm downfield shift for the signals of the methylene groups a, b and c (Scheme 1) of L in the presence of DP. The aromatic signals of the tpy moiety are almost not affected by DP coordination. The shifts observed in the presence of TP are lower, and almost negligible in the case of MP (see ESI†). Furthermore, the interaction of complex I at pH 7 with DP does not give significant changes in the UV-vis spectrum of the complex (see ESI†).
Of note, no interaction is detected by potentiometry between the complex [Zn(tpy)]2+ and MP and DP under our experimental conditions, while TP is very weakly bound (log K = 2.8 for the addition of P3O105− to the [Zn(tpy)]2+ complex). These results strongly suggest that the two protonated [9]aneN3 units play a relevant role in DP binding by the 1:1 Zn2+ complex of L, via the formation of hydrogen bonds and charge–charge interactions involving the protonated ammonium groups and the anion located in the cleft formed between the two facing macrocyclic units. In fact, DFT calculations carried out on the adduct formed between [ZnLH2(H2O)2]4+ and P2O74− with an implicit simulation of the aqueous environment suggest that DP is not directly bound to the metal center (see ESI†). For these calculations, the Zn2+ ion was considered pentacoordinate as normally found in its 1:1 complexes with tpy (see ESI†). In the optimized structure (Fig. 3) the two acidic protons are localized on secondary amine groups of two different [9]aneN3 moieties, in keeping with the higher basicity of secondary amine groups with respect to tertiary ones in H2O. This proton distribution also ensures a better minimization of the electrostatic repulsion between positive charges. More interestingly, the DP anion is encapsulated within the cleft delimited by the [9]aneN3 units, but it does not replace the water molecules in the coordination sphere of the metal. In fact, the P2O74− anion seems to prefer to give a hydrogen bonding network involving both the coordinated water molecules and the two ammonium groups of the receptor.
![Minimized structure of the adduct [ZnLH2(H2O)2(P2O7)].](/image/article/2012/CC/c1cc15934h/c1cc15934h-f3.gif) | ||
| Fig. 3 Minimized structure of the adduct [ZnLH2(H2O)2(P2O7)]. | ||
DP binding also affects the photophysical properties of the 1:1 Zn2+ complex of L. In fact, addition of DP to solutions of I at pH 7 leads to a progressive fluorescence quenching (Fig. 4) and the system is basically non-emissive in the presence of 1 equiv. of DP. Once again, this behavior can be reasonably ascribed to the formation of strong hydrogen bond contacts between the DP anion and the protonated ammonium functions of the receptor. In fact, as often observed in anion complexes with polyammonium receptors, hydrogen bonding is accompanied by a partial transfer of positive charge from the ammonium groups to the anion. The decreased charge on the [9]aneN3 units may allow the polyamine groups to undergo a photoinduced electron transfer process to the excited fluorophore, thus quenching the fluorescence emission.
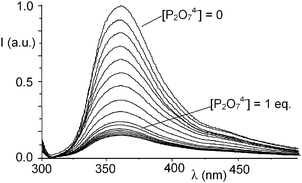 | ||
| Fig. 4 Fluorescence emission of complex I in H2O in the presence of increasing amounts of P2O74−. | ||
Of note, the emission of complex I is not affected by the presence of MP and only 8% reduced by a threefold excess of TP, confirming the weaker interactions of these anions with the metal-based receptor (see ESI†).
As a matter of fact, L represents a rare case of receptor which couples anion recognition and sensing, mainly thanks to an “allosteric effect” exerted by a metal cation. The Zn2+ ion, in fact, does not act as a direct anchoring site for DP through coordinative bonds, but induces a conformation change in the receptor structure, which allows substrate binding via weak, non-coordinative forces. The consequent new organization of the binding sites imparts the receptor selectivity in both anion binding and fluorescence sensing.
Financial support by MIUR (Project PRIN 2009 - 2009Z9ASCA) is gratefully acknowledged.
Notes and references
- J. L. Sessler, P. A. Gale and W. S. Cho, Anion Receptor Chemistry, Royal Society of Chemistry, Cambridge, 2006 CrossRef CAS; Supramolecular Chemistry of Anions, ed. A. Bianchi, K. Bowman-James and E. Garcia-España, Wiley-VCH, NY, 1997 CrossRef CAS; T. Gunnlaugsson, H. D. P. Ali, M. Glynn, P. E. Kruger, G. M. Hussey, F. M. Pfeffer, C. M. G. Santos and J. Tierney, J. Fluoresc., 2005, 15, 287 CrossRef CAS; C. Bazzicalupi, A. Bencini and V. Lippolis, Chem. Soc. Rev., 2010, 39, 3709 RSC; T. Sakamoto, A. Ojida and I. Hamachi, Chem. Commun., 2009, 141 RSC; L. Fabbrizzi, M. Licchelli, G. Rabaioli and A. Taglietti, Coord. Chem. Rev., 2000, 205, 85 CrossRef.
- D. L. Nelson and M. M. Cox, Lehninger Principles of Biochemistry, 5th edn, W. H. Freeman, NY, 2009 Search PubMed.
- Phosphorus in Environmental Technology—Principles and Applications, ed. E. Valsami-Jones, IWA Publishing, UK, 2004 Search PubMed.
- S. K. Kim, D. H. Lee, J.-I. Hong and J. Yoon, Acc. Chem. Res., 2009, 42, 23 CrossRef CAS; M. Kruppa and B. König, Chem. Rev., 2006, 106, 3520 CrossRef; I. Ravikumar and P. Ghosh, Inorg. Chem., 2011, 50, 4229 CrossRef; M. S. Han and D. H. Kim, Angew. Chem., Int. Ed., 2002, 41, 3809 CrossRef; P. Das, A. Ghosh, M. K. Kesharwani, V. Ramu, B. Ganguly and A. Das, Eur. J. Inorg. Chem., 2011, 3050 CrossRef; W.-H. Chen, Y. Xing and Y. Pang, Org. Lett., 2011, 13, 1362 CrossRef; J. Wen, Z. Geng, Y. Yin, Z. Zhang and Z. Wang, Dalton Trans., 2011, 40, 1984 RSC; J.-H. Lee, A.-R. Jeong, J.-H. Jung, C.-M. Park and J.-I. Hong, J. Org. Chem., 2011, 76, 417 CrossRef; Z. Zeng, A. A. Torriero, A. M. Bond and L. Spiccia, Chem.–Eur. J., 2010, 16, 9154 CrossRef; G. Ambrosi, M. Formica, V. Fusi, L. Giorgi, A. Guerri, E. Macedi, M. Micheloni, P. Paoli, R. Pontellini and P. Rossi, Inorg. Chem., 2009, 48, 5901 CrossRef; S. Khatua, S. H. Choi, H. Shin, J. Lee, K. Kim, Y. Do and D. G. Churchill, Inorg. Chem., 2009, 48, 2993 CrossRef; L. Rodriguez, J. C. Lima, A. J. Parola, F. Pina, R. Meitz, R. Aucejo, E. Garcia-España, J. M. Llinares, C. Soriano and J. Alarcon, Inorg. Chem., 2008, 47, 6173 CrossRef; H. N. Lee, Z. Xu, S. K. Kim, K. M. K. Swamy, Y. Kim, S.-J. Kim and J. Yoon, J. Am. Chem. Soc., 2007, 129, 3828 CrossRef; N. Shao, H. Wang, X.-D. Gao, R.-H. Yang and W.-H. Chan, Anal. Chem., 2010, 82, 4628 CrossRef; D. H. Lee, J. H. Im, S. U. Son, Y. K. Chung and J.-I. Hong, J. Am. Chem. Soc., 2003, 125, 7752 CrossRef.
- J. E. Coleman, Annu. Rev. Biochem., 1992, 61, 897 CrossRef CAS; W. Maret, BioMetals, 2001, 14, 187 CrossRef.
- C. Caltagirone, A. Mulas, F. Isaia, V. Lippolis, P. A. Gale and M. E. Light, Chem. Commun., 2009, 6279 RSC.
- C. Bazzicalupi, A. Bencini, C. Giorgi, B. Valtancoli, V. Lippolis and A. Perra, Inorg. Chem., 2011, 50, 7202 CrossRef CAS.
- C. Bazzicalupi, A. Bencini, A. Bianchi, A. Danesi, E. Faggi, C. Giorgi, S. Santarelli and B. Valtancoli, Coord. Chem. Rev., 2008, 252, 1052 CrossRef CAS.
- A. Bianchi and E. Garcia-España, J. Chem. Educ., 1999, 76, 1727 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details for ligand synthesis, potentiometric, UV-Vis, fluorescence, 1H NMR experiments and DFT calculations, stability constants and distribution diagrams for the complexes formed by L in the presence of Zn2+ and/or phosphate anions. See DOI: 10.1039/c1cc15934h |
| This journal is © The Royal Society of Chemistry 2012 |