Porous ‘Ouzo-effect’ silica–ceria composite colloids and their application to aluminium corrosion protection†
Martin J.
Hollamby
*,
Dimitriya
Borisova
,
Helmuth
Möhwald
and
Dmitry
Shchukin
*
Max Planck Institute of Colloids and Interfaces, Wissenschaftspark Potsdam-Golm, Am Muehlenberg 1, 14476 Potsdam, Germany. E-mail: hollamby.martinjames@nims.go.jp; dmitry.shchukin@mpikg.mpg.de
First published on 7th November 2011
Abstract
By exploiting spontaneous emulsification to prepare porous SiO2 particles, we report the formation of porous CeO2@SiO2 hybrid colloids and their incorporation into a silica–zirconia coating to improve the corrosion protection of aluminium.
The “Ouzo effect” is probably better known as a drinkers' conundrum than as a technique for the formation of colloidal particles. The careful addition of water to e.g. Pastis, Absinthe, etc. causes the spontaneous emulsification of a metastable droplet phase, driven by the aqueous insolubility of the flavour compound (trans-anethole). Several studies have sought to characterise the underlying process1 and to use it for the production of polymer particles and capsules.2 As a synthesis method, spontaneous emulsification has the advantage of requiring neither externally applied energy, nor stabilizing or templating species. Using a simple system consisting of water, ethanol, ammonia, hydrochloric acid and silicon alkoxides, porous silica particles were formed.3 However, few other reports use this method to form hard, porous colloidal particles and there have been no reports of their application. This is remarkable given that while porous particles have been developed for many applications (e.g. catalysis, drug delivery, chromatography, corrosion inhibition), almost all of the preparation methods suffer the requirement of templating species, high-temperature syntheses or high shear pre-emulsification.4–7 A spontaneous emulsification-based route to form porous materials would be cheap, simple, non-hazardous and therefore warrants further consideration. One way to functionalise the resultant particles (e.g.zeolite or silica) is to adsorb active nanoparticles.8 Such hybrid colloids would have the benefit of easy solution uptake and facile incorporation into e.g. coatings.
Here, a spontaneous emulsification process was used to prepare porous silica particles,3 which are capable of nanoparticle and/or small molecule uptake. Ceria was chosen as the active content, given its known use both in oxidation catalysis4,9 and in corrosion protection.10–12 Several routes exist to form ceria nanocrystals.9,13 Here, an adapted precipitation method was employed,14 due to its simplicity and high yield. Fig. 1a shows the characterization of the ceria particles by XRD. Peaks corresponding to the fluorite ceria structure are clearly noted and marked, with substantial broadening indicating the presence of nano-crystalline grains. The minimum grain size was calculated to be 5.5 nm by the Scherrer equation15 using shape factor = 0.9, in line with an approximate particle size estimated by TEM (d ≈ 5 nm, Fig. S1, ESI†). TEM images of the silica spheres formed by spontaneous emulsification are shown in Fig. 1b. The sample mainly consisted of polydisperse porous particles with dmean ≈ 200 nm (see ESI†). The dependence on the zeta potential with pH (Fig. 1c) for the silica and ceria particles showed typical isoelectric points (IEP) at pH 3.2 and pH 8.5, respectively.16 The BET surface area of the SiO2 particles was calculated by N2 sorption (Fig. 1d) to be 128 m2 g−1, with a BJH desorption pore volume of 0.38 ml g−1 and pore diameters in the range 3–20 nm (dmean = 10 nm).
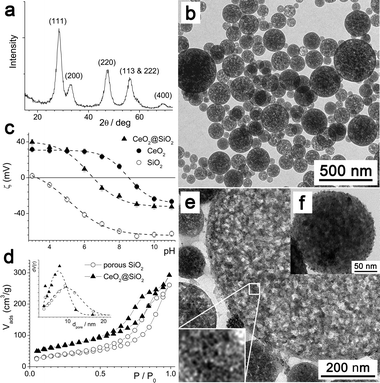 | ||
| Fig. 1 (a) XRD spectrum of CeO2 particles. (b) TEM image of the porous silica particles. (c) Zeta potential, ζ, vs. pH for the different particles. (d) N2 sorption data and (inset) pore size distributions for the different particles. (e) TEM image of a microtomed section of the CeO2@SiO2 particles. (f) Standard TEM image of a CeO2@SiO2 particle. | ||
In the region pH 3.2–8.5 the silica and ceria particles have opposing surface charges (cf.Fig. 1c) and therefore adsorption of ceria to the silica surface was expected.17 Upon mixing solutions of the particles, a light yellow solution was formed. Adsorption was evidenced by a shift in IEP to pH 6.6 and was detected by DLS (see ESI†) and TEM (Fig. 1e and f). Inside the SiO2 spheres (see ESI†), darker spots can be seen (e.g. expanded section, Fig. 1e), which are interpreted as being embedded ceria particles. N2 sorption indicated a narrower pore size distribution (3–13 nm, dmean = 7 nm—see inset, Fig. 1d), suggesting that ceria particles have entered the larger pores. The CeO2@SiO2 particles have an increased BET surface area of 224 m2 g−1, but a similar total pore volume of 0.36 ml g−1, suggesting additional roughening and pore formation by adsorbed and sequestered CeO2.
One potential application of these particles is as hard-shell nano-reservoirs into which corrosion inhibitors could be sequestered. Corrosion protection is an important issue in which any improvement can lead to significant economic benefits. Corrosion inhibitor-containing capsules dispersed throughout a passive coating matrix have been reported that provide protection to both aluminium AA2024-T319 and galvanized steel.6Inhibitor release is thought to be directed by the change in local electrochemical potential (e.g. local pH) around the corrosion site.6,20 To complement CeO2 corrosion inhibition, 8-hydroxyquinoline (8HQ) was used, which is an effective anodic inhibitor that has exhibited synergistic effects with Ce-containing compounds.5,118HQ adsorption to the CeO2@SiO2 particles was visualized via a colour change in the particles to red-brown (Fig. 2a and b), indicative of Ce–8HQ complex formation.21
![(a, b) Visual appearance of the CeO2@SiO2 particle solutions after pelleting by centrifugation (a) before and (b) after 8HQ adsorption. (c) Release curves from the 8HQ + CeO2@SiO2 particles as a function of time and solution pH. Fractional release, ΔR, is given by [8HQ]t/[8HQ]max. Lines are fits to a modified Korsmeyer–Peppas model ΔR = ktn + a.18](/image/article/2012/CC/c1cc15992e/c1cc15992e-f2.gif) | ||
| Fig. 2 (a, b) Visual appearance of the CeO2@SiO2 particle solutions after pelleting by centrifugation (a) before and (b) after 8HQ adsorption. (c) Release curves from the 8HQ + CeO2@SiO2 particles as a function of time and solution pH. Fractional release, ΔR, is given by [8HQ]t/[8HQ]max. Lines are fits to a modified Korsmeyer–Peppas model ΔR = ktn + a.18 | ||
After rigorous, the release kinetics of 8HQ from CeO2@SiO2 was assessed at different solution pH (see ESI†). The results are presented in Fig. 2b as fractional release vs. time and in all cases, a strongly pH dependent burst release is noted, followed by a slower discharge. Release occurs more quickly at pH 1 and pH 12 than at pH 5 or pH 9. The data were fitted to a modified Korsmeyer–Peppas model,18 where ΔR is the fractional release at time, t in minutes, k is a rate constant, a is the fraction released in the burst and n gives mechanistic insight.22 At pH 1 and 12, both a and k are greater than at pH 5 and 9 (Table S1, ESI†). In all cases, n < 0.5, indicating a quasi-Fickian diffusion process. While neutral pH was not tested due to experimental difficulties, both the burst magnitude and release rate are likely to be low. For corrosion applications, the initial burst is particularly important as it describes the amount of inhibitor available on contact with a corrosive solution e.g. in a scratch. As corroding areas have high acidity and basicity at the anode and cathode, respectively, the larger release at pH extremes is desirable. On the other hand, the reduced release around neutrality ought to limit the leeching of inhibitor from the coating. While they describe the release of inhibitor in solution, the results do suggest that the hybrid particles could act as an effective delivery system.
To further test this, the effect of including the particles into a silica–zirconia sol–gel coating19 on its anti-corrosion performance was assessed. This study investigates for the first time the effect of incorporating two known inhibitors into the same coating. Samples tested include uncoated aluminium and aluminium dip-coated with the sol–gel matrix alone (control, SG) and the sol–gel matrix incorporating bare SiO2 particles (SiO2 + SG), CeO2@SiO2 particles (CeO2@SiO2 + SG), SiO2 with adsorbed 8HQ (8HQ,SiO2 + SG) and CeO2@SiO2 with adsorbed 8HQ (8HQ,CeO2@SiO2 + SG). In all cases, the concentration of nanoparticles was 1.3 wt% in the coating. Fig. 3(a–f) shows images of corrosion damage to the samples after immersion in 1 M NaCl. Clear improvements in protection can be observed for all of the coatings containing particles. While signs of corrosion are visible in all cases, SiO2 + SG and CeO2@SiO2 + SG seem to provide the best protection, with fewer pits. To quantify this, electrical impedance spectroscopy (EIS) was used. From the measured phase shift and absolute impedance, separate contributions from resistance, R, and capacitance, C, at different frequencies, f, were calculated (see ESI†), as shown in Fig. 3(g and h) (5 days immersion). The plot of absolute impedance and phase shift vs. frequency is in the ESI† (Fig. S2). All of the data is consistent with a breached coating and a corroding sample, but there are differences in performance. In general, the observed values at high frequency (103–105 Hz) are most likely to reflect the properties of the coating, whereas those at low frequency (10−2–10−1 Hz) probably reflect the properties of the double layer at the metal surface. Lower values of C can imply a smaller affected area, a wider charge separation or a weaker dielectric within the capacitor. Given that all coatings were made in the same way and include only 1.3 wt% added material, both the thickness and the dielectric will be similar, so the differences in C at high frequency are likely to be due to differences in the intact area. The addition of particles appears to generally result in a more intact coating, with a lower capacitance as noted before,23 which probably also explains the higher R values at 103–105 Hz. However, adding 8HQ leads to a decrease in R and an increase in C vs. the bare samples, suggesting that the coating is more porous. A similar pattern is noted in the lower frequency region, with all coatings containing particles exhibiting lower C and higher R than the control. Again, it is likely to be the double-layer area (i.e. number of pits) that is responsible for the differences in the values. Here the inclusion of the CeO2@SiO2 particles provides the best performance, possibly due to some cathodic inhibition in addition to increased barrier integrity. While no strong anodic inhibition from the 8HQ content is detected, the gaps between values of C, R for samples with or without 8HQ become smaller at lower frequency, which might be an indirect measurement of an inhibitive effect.
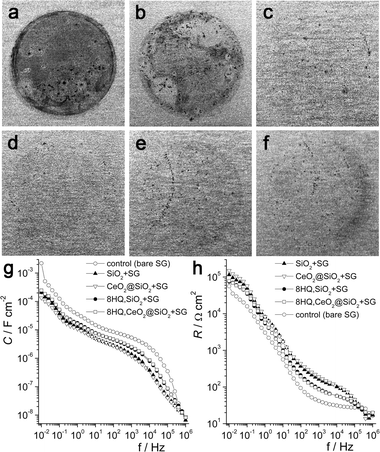 | ||
| Fig. 3 (a) Photograph of the bare AA2024 substrate after 3 days immersion in 1 M NaCl, after rinsing with water. (b–f) Images of the coated samples after 7 days immersion in 1 M NaCl, after rinsing with water. Key: b: control/bare sol–gel (SG), c: SiO2 + SG, d: CeO2@SiO2 + SG, e: 8HQ,SiO2 + SG, f: 8HQ,CeO2@SiO2 + SG. (g) Capacitance vs. frequency for the coated Al substrates after 5 days. (h) Resistance vs. frequency for the samples after 5 days. | ||
To better assess the deterioration in corrosion protection, the change in absolute impedance at 0.1 Hz (Zabs, 0.1 Hz) with immersion time can be used (Fig. S3, ESI†). For all coated substrates, Zabs,0.1 Hz decreases over the course of the measurement and after just one day, the protection afforded by the control has stabilised at a low level. The SiO2 + SG and CeO2@SiO2 + SG samples provide better protection for longer times, with a constant value reached only near the end of the test. This probably reflects the ability of the particles to strengthen intra-coating bonding by acting as nucleation points or bridging agents during the formation process. While the initial protection is lower than the control for coatings incorporating 8HQ, the rate in decrease is less and improved impedance is seen after immersion for just one day. This might be explained as follows: samples including 8HQ provide lower initial barrier protection due to inference with the gelling and curing processes, leading to the formation of more micro-cracks or ion diffusion pathways in the coating. However, after the onset of corrosion, available inhibitor is guided through these cracks to the interface by changes in the local pH, increasing the impedance of the double layer. After 7 days immersion, Zabs,0.1 Hz is quite similar for all of the samples incorporating particles. Significantly, the values are 2–3 times higher than that for the control and 4–5 times higher than that of the bare substrate, indicating better corrosion protection.
In summary, we report a method to spontaneously form hybrid nano-structures consisting of ceria nanoparticles supported on a porous silica colloid. The structures are characterized by a high surface area and can be used to adsorb useful molecules (e.g.8HQ) for future release. The particles were successfully included into a silica–zirconia sol–gel coating and improved protection versus the control coating was noted in all cases. If they were to be dispersed into an industrial water-based coating matrix they would certainly aid its barrier properties and could form the basis of a successful self-healing anticorrosion coating. Spontaneous emulsification, which requires neither template nor externally applied energy, has been shown to be a useful greener method to generate silica nanostructures that could find future use either in corrosion prevention or, perhaps in other fields, as e.g. an easily recoverable catalyst support.
We would like to thank Rona Pitschke and Heike Runge for help with TEM, Alex Kraupner for N2 sorption, Zoe Schnepp for XRD and Heidi Zastrow for Zeta potential measurements. The work was financially supported by ForMat program of the German Ministry for science and education (BMBF) and EU FP7 MUST project.
Notes and references
- (a) S. A. Vitale and J. L. Katz, Langmuir, 2003, 19, 4105–4110 CrossRef CAS; (b) I. Grillo, Colloids Surf., A, 2003, 225, 153–160 CrossRef CAS; (c) D. Carteau, D. Bassani and I. Pianet, C. R. Chim., 2008, 11, 493–498 CrossRef CAS.
- F. Ganachaud and J. L. Katz, ChemPhysChem, 2005, 6, 209–216 CrossRef CAS.
- W. T. Minehan and G. L. Messing, Colloids Surf., 1992, 63, 181–187 CrossRef CAS.
- B. Puertolas, B. Solsona, S. Agouram, R. Murillo, A. M. Mastral, A. Aranda, S. H. Taylor and T. Garcia, Appl. Catal., B, 2010, 93, 395–405 CrossRef CAS.
- I. Kartsonakis, I. Daniilidis and G. Kordas, J. Sol-Gel Sci. Technol., 2008, 48, 24–31 CrossRef CAS.
- M. J. Hollamby, D. Fix, I. Dönch, D. Borisova, H. Möhwald and D. Shchukin, Adv. Mater., 2011, 23, 1361–1365 CrossRef CAS.
- B. L. Cushing, V. L. Kolesnichenko and C. J. O'Connor, Chem. Rev., 2004, 104, 3893–3946 CrossRef CAS.
- (a) K. Gude and R. Narayanan, J. Phys. Chem. C, 2011, 115, 12716–12725 CrossRef CAS; (b) P. Sonström, M. Adam, X. Wang, M. Wilhelm, G. Grathwohl and M. Bäumer, J. Phys. Chem. C, 2010, 114, 14224–14232 CrossRef CAS; (c) A. Dong, Y. Wang, Y. Tang, N. Ren, Y. Zhang and Z. Gao, Chem. Mater., 2002, 14, 3217–3219 CrossRef CAS.
- A. Trovarelli, Catalysis by Ceria and Related Materials, Imperial College Press, London, 2002, vol. 2 Search PubMed.
- R. Di Maggio, S. Rossi, L. Fedrizzi and P. Scardi, Surf. Coat. Technol., 1997, 89, 292–298 CrossRef CAS.
- S. Szunerits and D. R. Walt, Anal. Chem., 2002, 74, 886–894 CrossRef CAS.
- N. C. Rosero-Navarro, S. A. Pellice, A. Durán, S. Ceré and M. Aparicio, J. Sol-Gel Sci. Technol., 2009, 52, 31–40 CrossRef CAS.
- M. J. Hollamby, K. Trickett, A. Vesperinas, C. Rivett, D. C. Steytler, Z. Schnepp, J. Jones, R. K. Heenan, R. M. Richardson, O. Glatter and J. Eastoe, Chem. Commun., 2008, 5628–5630 RSC.
- T. Taniguchi, T. Watanabe, N. Sakamoto, N. Matsushita and M. Yoshimura, Cryst. Growth Des., 2008, 8, 3725–3730 CrossRef CAS.
- A. L. Patterson, Phys. Rev., 1939, 56, 978 CrossRef CAS.
- M. Kosmulski, Chemical properties of material surfaces, Marcel Dekker, New York, 2001 Search PubMed.
- K. Esumi, H. Idogawa and K. Meguro, Bull. Chem. Soc. Jpn., 1988, 61, 2287–2290 CrossRef CAS.
- X. Huang and C. S. Brazel, J. Controlled Release, 2001, 73, 121–136 CrossRef CAS.
- D. G. Shchukin and H. Möhwald, Small, 2007, 3, 926–943 CrossRef CAS.
- D. Fix, E. V. Skorb, D. G. Shchukin and H. Möhwald, Meas. Sci. Technol., 2011, 22, 075704 CrossRef.
- W. Westwood and A. Mayer, Analyst, 1948, 73, 275 RSC.
- S. C. Basak, K. S. Kumar and M. Ramalingam, Rev. Bras. Cienc. Farm., 2008, 44, 477–483 Search PubMed.
- J. Malzbender and G. de With, Adv. Eng. Mater., 2002, 4, 296–300 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental detail, supporting figures, table of fitted parameters for the release experiment. See DOI: 10.1039/c1cc15992e |
| This journal is © The Royal Society of Chemistry 2012 |