Visible light-driven CO2 reduction by enzyme coupled CdS nanocrystals†‡
Yatendra S.
Chaudhary
ad,
Thomas W.
Woolerton
a,
Christopher S.
Allen
b,
Jamie H.
Warner
b,
Elizabeth
Pierce
c,
Stephen W.
Ragsdale
c and
Fraser A.
Armstrong
*a
aInorganic Chemistry Laboratory, Department of Chemistry, University of Oxford, South Parks Road, Oxford OX1 3QR, United Kingdom. E-mail: fraser.armstrong@chem.ox.ac.uk; Tel: +44-1865 272647
bDepartment of Materials, University of Oxford, 16 Parks Road, Oxford OX1 3PH, United Kingdom
cDepartment of Biological Chemistry, University of Michigan, Ann Arbor, Michigan 48109-0606, USA
dColloids and Materials Chemistry Department, Institute of Minerals and Materials Technology (CSIR), Bhubaneswar 751013, India
First published on 15th November 2011
Abstract
Assemblies of carbon monoxide dehydrogenase molecules with CdS nanocrystals show fast CO2 reduction driven by visible light. Activity is strongly influenced by size and shape of nanocrystals, and by the nature of the electron donor.
Driven by environmental and long term energy-security, but also by intellectual challenges, there is a thriving interest in innovating ‘bottom-up’ photoelectrochemical devices to produce fuels using energy from sunlight. The essential parts of a physical solar fuel synthesiser (an artificial photosynthesis device) are a light-capturing component to generate the electron-hole separation and electrocatalytic centres for converting electrons and holes into chemical energy stores—the fuel and oxidised waste product. A good catalyst should produce a single chemical product at high rates that compete with electron-hole recombination. Although the most obvious fuel is H2 from water, there is also much interest in converting concentrated sources of CO2.
A number of electrocatalysts are currently known to mediate CO2 reduction, including Pd and Re complexes as well as the simple pyridinium ion.1–8 However, progress is limited by low turnover frequencies and/or high overpotential requirements. The ultimate source of the problem is that reduction of CO2 in one-electron steps via the intermediate free radical CO2˙− incurs a large overpotential cost (>1 V) whereas a perfect catalyst would overcome this barrier with well orchestrated proton-coupled electron transfers to tightly bound intermediates.
Despite their size, many enzymes are excellent electrocatalysts.9Carbon monoxide dehydrogenases (CODHs) adsorbed on an electrode catalyse the electrochemical interconversion of CO2 and CO with high turnover rates at minimal overpotentials.10 The active site of CODHs from anaerobic organisms is a [Ni4Fe-4S] centre known as the C cluster. Recently, we reported how CODH I (henceforth CODH) from Carboxydothermus hydrogenoformans, when attached to titanium oxide (P25) nanoparticles, converts CO2 to CO under visible light illumination.11 Since TiO2 is a wide band gap semiconductor (Eg = 3.1 eV), the TiO2 particles were photosensitised by co-attachment of a visible light harvesting Ru complex [RuII(bpy)2(4,4′-(PO3H2)2-bpy)]2+. The hole left on the Ru complex was quenched by a sacrificial electron donor. We have since sought to simplify this system, retaining the highly active enzyme electrocatalyst but looking for alternative light-capturing components. Semiconductor quantum dots and nanorods offer interesting possibilities because the reducing and photo-generated charge carrier separation efficacy of semiconductors strongly depends upon size quantisation effects. Such properties can thus be tuned by varying the size and shape of nanomaterials. In this Communication, we report the assembly of CODH on visible light harvesting CdS nanocrystals with different size and shape to eliminate the use of dye (Fig. 1). The activity for CO2 reduction to CO under visible light, the role of sacrificial agents and particle (CdS nanocrystal) size and shape is discussed. The conduction band (CB) edge of bulk CdS lies at ECB = −0.87 V vs.SHE (pH 6)—significantly more negative than that of TiO2—and CdS supports are therefore able to provide plenty of driving force to reduce CO2 to CO (reduction potential −0.46 V vs.SHE, at pH 6).
The detailed method of the synthesis of CdS quantum dots (QDs) is reported elsewhere.12 Typically, a homogeneous mixture of cadmium acetate, dodecylamine and sulfur powder was prepared and transferred to a Teflon-lined stainless steel pressure vessel. It was heated at 220 °C for 10 h. The CdS sample was then collected by centrifugation, repeatedly washed with ethanol and carbon disulfide and dried at room temperature under ambient conditions. The CdS powder thus obtained was yellow in colour. The broad peaks shown in the X-ray diffraction patterns of CdS powder samples (supporting information, Fig. S1‡), suggest the formation of ultra-small particles. Indexing of these peaks [(111), (220) and (311)] reveals the formation of cubic phase. To establish the size and morphology of the CdS particles, transmission electron microscopy (JEOL 4000EX operated at 80 kV) was used. The TEM image shown in Fig. 2A indicates formation of quantum dots with dimensions 5.8 ± 1.8 nm, mostly spherical in shape. As the band gap energy is an important parameter for the harvesting of solar radiation, the UV-visible absorbance spectrum was recorded (Perkin Elmer Lambda 19). The band gap, estimated from the absorption data, is of the order of 2.3 eV (supporting information, Fig. S2‡) thus suggesting the suitability of CdS QDs for harvesting visible solar radiation. In addition, CdS nanorods (NRs, also yellow) with average dimensions of 42 ± 10 nm in length and 10 ± 1 nm in width were synthesised (Fig. 2C), following a reported procedure.13
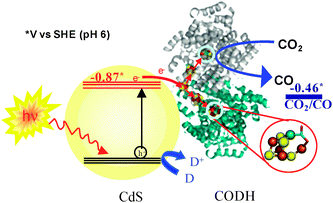 | ||
| Fig. 1 Schematic representation of visible light-driven CO2 reduction using CODH-CdS nanocrystal assemblies. D represents an electron donor. | ||
 | ||
| Fig. 2 TEM images of CdS nanocrystals. (A) as-synthesized QDs, (B) after calcination and (C) as-synthesized nanorods. | ||
In all cases co-attachment of CODH (CODH I prepared as before11) and CdS nanocrystals was carried out as follows. As-synthesized CdS nanocrystals were exposed to UV light and thoroughly washed with ethanol, before co-attaching CODH with CdS nanocrystals so as to ensure an efficient interfacial exciton transport. CdS nanocrystal powder (10 mg) was dispersed in 5 ml of the desired aqueous buffer (pH 6) in a pressure vessel and sonicated for 20 min. to ensure formation of a stable suspension. Then 13.9 μl of 184 μM CODH (2.56 nmol) was introduced to the CdS suspension, followed by gentle stirring for 20 min. to allow co-attachment. Due to the O2-sensitivity of the enzyme, all manipulations were performed in a glove box (Belle Technologies, O2 < 3 ppm) under a N2 atmosphere. The amount of CODH co-attached with CdS quantum dots (CODH-QDs) or with CdS nanorods (CODH-NRs) was determined by comparing the absorbance at 280 nm of the solution before and after mixing CODH with nanocrystals. The supernatant was obtained by centrifugation followed by filtration (supporting information, Fig. S3‡). Typically, 10 mg of CdS QDs co-attached with 0.59 ± 0.32 nmol CODH, and 10 mg of CdS NRs co-attached with <0.1 nmol CODH.
For CO2 photo-reduction, pressure vessels containing CODH-CdS nanocrystal assemblies were sealed with rubber septa and purged with 98% CO2/2% CH4 for 20 min. while stirring. The vessel was then exposed to a light source (Kodak Carousel S-AV 1010 projector fitted with a 250 W tungsten-halogen bulb and a 420 nm long-pass filter, UQG Optics). The light intensity incident on CODH-CdS nanocrystal assemblies was ca. 23 mW cm−2, measured using a power/energy meter (Fisher Scientific, FB61163), and to minimise heating, the vessel was immersed in a water bath at 20 °C throughout the CO2 reduction experiments. Production of CO was measured at regular time intervals by gas chromatography (Agilent Technologies, 7890A GC equipped with Restek ShinCarbon packed column) using He carrier gas and a thermal conductivity detector. The amount of CO was quantified against the internal standard (2% CH4) with respect to calibration plots obtained with various known amounts of CO (supporting information, Fig. S4‡).
The progress of CO production at CODH-QDs using different electron donors is shown in Fig. 3. In all cases, 0.35 M 2-(N-morpholino)ethanesulfonic acid (MES) was used to buffer the acidifying effect of CO2 in solution: MES is also able to act as the electron donor, quenching photo-generated holes formed in the CdS QDs. Additional sacrificial components had either negligible or detrimental effects on the rate of CO production. Additional ascorbate (0.2 M) or KI (0.3 M) suppressed all detectable activity, and TEOA (0.2 M) decreased activity by around 80%. The reason behind the variable activity shown by different electron donors is unclear at this point, although the affinity of –COOH or –NH2groups for CdS surfaces is well established.12,14 Therefore, it is likely that the variation in activity is related to the different degrees of interaction (binding) of these electron donors with the CdS surface. In separate experiments, the concentration of MES was varied between 0.2–0.5 M but there was no significant change in CO production (supporting information, Fig. S5‡).
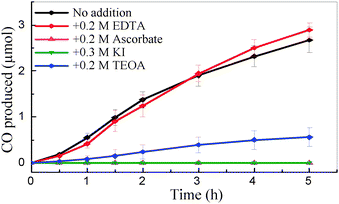 | ||
| Fig. 3 CO produced versus time by CODH-QDs with various electron donors (pH 6.0, 20 °C). All solutions contained 0.35 M MES. | ||
Furthermore, the photocatalytic activity of such hybrid assemblies is highly susceptible to the surface characteristics of each component, since this affects the electroactive co-attachment of the enzyme, and the efficiency of electron transfer. In the case of the CdS moiety, the size of the particle affects the nature of the surface, as well as the light harvesting characteristics. To investigate size effects, the CdS QDs were subjected to calcination at 450 °C for 45 min. (heated from room temperature at 5 °C min−1). Upon heat treatment, the CdS QDs converted into larger particles/clusters of irregular shape (Fig. 2B). The calcined CdS particles were co-attached with CODH using the previously described method to form CODH-CdScalc, and CO2 photo-reduction experiments were subsequently performed. In contrast to the CO2 photo-reduction activity shown by CODH-QDs, CODH-CdScalc showed no detectable CO formation, even after 5 h of irradiation (Fig. 4A). To explore the factors responsible for the complete loss of activity, UV-vis measurements were firstly made, to determine the amount of CODH co-attached with calcined CdS particles (using the same method as described for QDs and NRs). This showed co-attachment of CODH with calcined CdS particles, although the amount of co-attached CODH (0.2 ± 0.1 nmol) was lower than for CODH-QDs (0.59 nmol). Secondly, the XRD pattern recorded for the CdS particles after calcination shows no change in phase, which remains entirely cubic (supporting information, Fig. S1‡). The loss of activity thus appears to relate to particle size. The larger particles have more grain boundaries than quantum dots; grain boundaries act as recombination sites for photo-generated charge carriers.
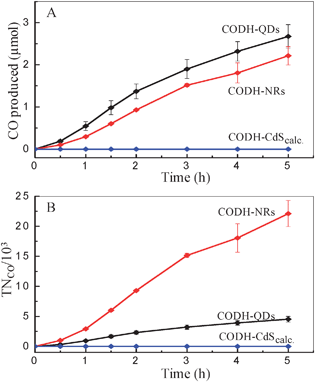 | ||
| Fig. 4 (A) Production of CO and (B) turnover number, versus time at different CODH-CdS assemblies. (0.35 M MES, pH 6.0, 20 °C). | ||
To evaluate the effect of shape of CdS nanocrystals on activity, CO2 photo-reduction experiments with CODH-NRs were performed, using 0.35 M MES as electron donor. Although the amount of CODH co-attached to NRs is very low (<0.1 nmol) significant production of CO was observed (Fig. 4A). Turnover numbers (TNCO, per molecule of co-attached enzyme) are shown in Fig. 4B. The CODH-NRs assemblies showed an average turnover frequency of 1.23 s−1 per enzyme molecule, compared to 0.25 s−1 for the CODH-QDs sample. Although there is a ca. 0.5 s−1 error, 1.23 s−1 compares well with other photocatalytic activities for CO2 photo-reduction reported using semiconductor or supramolecular photocatalysts. Some of the highest turnover frequencies reported for CO2 photo-reduction in the literature include 0.0065 s−1 for CO production using a [RuRe(P(OEt)3)]3+ supramolecular catalyst15 and 0.14 s−1 for CO production using CODH on Ru-dye sensitised TiO211 (literature data converted to common units).
From the relative radii of CODH and CdS QDs (the hydrodynamic diameter of CODH is approximately 10 nm, double the size of CdS QDs)16 we estimate that up to 10 CdS QDs can bind to each enzyme molecule in close contact, assuming that all CdS QDs are spherical in shape. The binding interactions are unclear but probably comprise both hydrophobic and polar contacts. The quantum efficiency is low-around 10−4 for both CODH-QDs and CODH-NRs, but this is not very meaningful because the light source supplies photons of all wavelengths greater than 420 nm, and only a fraction of these are useful for CdS bandgap excitation.
Our interest has focussed on the turnover frequency per CODH catalyst, which is greater than the rate of synthetic catalysts but remains considerably lower than the maximum capability of the enzyme.17 As we have described, variations in size and shape of CdS nanocrystals and type of electron donor have significant effects on the activity. These observations, including the higher rate of CO2 photo-reduction observed with NRs (lower exciton recombination probability due to lower dimensionality)18 strongly suggest that recombinations of photogenerated charge carriers are limiting the rate of CO production.
Yatendra Chaudhary acknowledges the financial support provided by the European Commission under the Marie Curie Programme (PIIF-GA-2009-253602). We also thank the EPSRC-Supergen 5 program (FAA), BBSRC-Grant H003878-1 (FAA) and NIH - GM39451 (SWR) for support throughout this work.
Notes and references
- J. Hawecker, J. M. Lehn and R. Ziessel, Chem. Commun., 1984, 328 RSC
.
- S. Meshitsuka, M. Ichikawa and K. Tamaru, Chem. Commun., 1974, 158 RSC
- C. M. Lieber and N. S. Lewis, J. Am. Chem. Soc., 1984, 106, 5033 CrossRef CAS
- S. Slater and J. H. Wagenknecht, J. Am. Chem. Soc., 1984, 106, 5367 CrossRef CAS
- D. L. Delaet, R. Delrosario, P. E. Fanwick and C. P. Kubiak, J. Am. Chem. Soc., 1987, 109, 754 CrossRef CAS
- D. L. Dubois, Comments Inorg. Chem., 1997, 19, 307 CAS
- J. M. Smieja and C. P. Kubiak, Inorg. Chem., 2010, 49, 9283 CrossRef CAS
- E. B. Cole, P. S. Lakkaraju, D. M. Rampulla, A. J. Moris, E. Abelev and A. B. Bocarsly, J. Am. Chem. Soc., 2010, 132, 11539 CrossRef
- F. A. Armstrong and J. Hirst, Proc. Nat. Aca. Sci., 2011, 108, 1404 Search PubMed
- A. Parkin, J. Seravalli, K. A. Vincent, S. W. Ragsdale and F. A. Armstrong, J. Am. Chem. Soc., 2007, 129, 10328 CrossRef CAS
- T. W. Woolerton, S. Sheard, E. Reisner, E. Pierce, S. W. Ragsdale and F. A. Armstrong, J. Am. Chem. Soc., 2010, 132, 2132 CrossRef CAS
- Y. Cheng, Y. Wang, F. Bao and D. Chen, J. Phys. Chem. B, 2006, 110, 9448 CrossRef CAS
- J. Joo, H. B. Na, T. Yu, J. H. Yu, Y. W. Kim, F. Wu, J. Z. Zhang and T. Hyeon, J. Am. Chem. Soc., 2003, 125, 11100 CrossRef CAS
- J. G. C. Veinot, J. G. Galloro, L. Pugliese, R. Pestrin and W. J. Pietro, Chem. Mater., 1999, 11, 642 Search PubMed
- S. Sato, K. Koike, H. Inoue and O. Ishitani, Photochem. Photobiol. Sci., 2007, 6, 454 RSC
- H. Dobbek, V. Svetlitchnyi, L. Gremer, R. Huber and O. Meyer, Science, 2001, 293, 1281 CrossRef CAS
- V. Svetlitchnyi, C. Peschel, G. Acker and O. Meyer, J. Bacteriol., 2001, 183, 5134 CrossRef CAS
- Q. Zhang and G. Cao, Nanotoday, 2011, 6, 91 CrossRef CAS
Footnotes |
| † This article is part of the ChemComm ‘Artificial photosynthesis’ web themed issue. |
| ‡ Electronic supplementary information (ESI) available: The XRD pattern for CdS QDs, UV-vis absorption spectrum of CdS QDs, UV-vis absorption spectra showing the co-attachent of CdS QDs with CODH and the procedure, calibration plot for GC quantification of CO, and CO2 photo-reduction data for CODH-QDs assemblies with the variation of MES concentration. See DOI: 10.1039/c1cc16107e |
| This journal is © The Royal Society of Chemistry 2012 |