Catalytic application of shape-controlled Cu2O particles protected by Co3O4 nanoparticles for hydrogen evolution from ammonia borane†
Yusuke
Yamada
*a,
Kentaro
Yano
a and
Shunichi
Fukuzumi
*ab
aDepartment of Material and Life Science, Division of Advanced Science and Biotechnology, Graduate School of Engineering, Osaka University, ALCA, Japan Science and Technology Agency (JST), Suita, Osaka 565-0871, Japan. E-mail: yamada@chem.eng.osaka-u.ac.jp; fukuzumi@chem.eng.osaka-u.ac.jp; Fax: +81-6-6879-7370; Tel: +81-6-6879-7368
bDepartment of Bioinspired Science, Ewha Womans University, Seoul, 120-750, Korea
First published on 11th November 2011
Abstract
Cu2O particles are active catalysts for hydrogen evolution from ammonia borane (AB) by hydrolysis, however, Cu2O particles easily form agglomerates as a result of highly reduced conditions during the reaction. In order to suppress agglomerate formation, capping of Cu2O with organic reagents or inorganic materials was performed and the catalytic reactivity in AB hydrolysis was examined. Among the examined methods, capping of Cu2O particles with Co3O4 nanoparticles was the most effective to avoid agglomerate formation of Cu2O particles. The finding enabled us to examine the shape effect of Cu2O particles on the catalytic reactivity in AB hydrolysis in the presence of Co3O4 nanoparticles. Comparisons of turnover frequencies for hydrogen evolution of Cu2O–Co3O4 composites, in which Cu2O particles were in the shape of 50-facets, cube, octahedron or rhombicuboctahedron, indicated that the composite with Cu2O with the shape of 50-facets showed more than 7-fold higher hydrogen evolution rate normalized by surface area than the composite with Cu2O with the octahedral shape. The size and shape effects of Co3O4 nanoparticles were also investigated on their ability to protect Cu2O from agglomeration. Comparisons of the catalytic reactivity of Cu2O particles decorated with Co3O4 nanoparticles of different sizes and shapes in terms of amounts and rates of hydrogen evolved by AB hydrolysis indicated that the size of Co3O4 nanoparticles is more important than the shape to exhibit high catalytic reactivity.
Broader contextHydrogen is regarded as a potential clean fuel for the next generation together with PEM fuel cells. In order to transport hydrogen as a fuel for mobile uses, hydrogen storage is very critical because hydrogen has poor energy content per volume. Storage of hydrogen in ammonia borane could be a solution because ammonia borane contains relatively high hydrogen contents (19.6%) and easily releases hydrogen under aqueous conditions with an appropriate catalyst. Among non-precious metals, Cu2O shows the high catalytic reactivity toward ammonia borane hydrolysis, however, severe deactivation has been observed due to agglomeration. Here we show a novel but conventional method to suppress the deactivation of Cu2O by decorating Cu2O particles partly with nanoparticles of Co3O4. The Co3O4 particles protect the Cu2O particles from agglomeration, resulting in high catalytic reactivity of Cu2O–Co3O4 composites. |
1. Introduction
Hydrogen is regarded as a promising fuel in the future because of its high energy content per mass, which is higher than petroleum. Thus, hydrogen has been used for running a proton exchange membrane fuel cell, PEMFC, which increases energy efficiency over internal combustion engines and decreases formation of harmful chemicals such as SOx, NOx, CO2 or particulate matters.1–3 A critical drawback of hydrogen is obviously its poor volumetric energy density.4,5Hydrogen evolution from liquid or solid chemicals on-site is suitable for application in transportation.6 Chemical hydrides are solid materials attracting a great deal of attention owing to their high gravimetric and volumetric storage capacity of hydrogen.7 Among the chemical hydrides, the ammonia borane complex, NH3BH3 (AB), is a promising candidate, because AB contains 19.6 wt% of hydrogen and more importantly this is stable in an aqueous solution at neutral pH as well as in the solid state.8Various types of metals and metal complexes have been reported to catalyse hydrogen evolution by thermal decomposition9,10 or solvo- or hydrolytic decomposition of ammonia borane.11–27 For the hydrolytic decomposition, precious metals such as Pt and Rh have been employed as active catalysts.8,28–30 However, industry demands more active catalysts made of abundant resources for a practical use. Among non-precious metals, copper catalysts such as Cu2O have been reported to act as catalysts for hydrolytic decomposition of AB although their activities are moderate compared with precious metal catalysts.8,31–34 For improvement of the catalytic behaviour of Cu2O particles, size and shape effects of Cu2O should be clarified. However, Cu2O particles easily form agglomerates leading to the loss of the catalytic reactivity under reductive conditions. Protection of Cu2O particles is necessary for evaluation of the shape effect of Cu2O particles on catalysis under reductive conditions.35,36
In general, covering surfaces of metallic particles with organic reagents or inorganic materials is a promising method to protect metallic particles from agglomeration. For example, an organic capping reagent of poly-N-vinyl-2-pyrrolidone (PVP) has been used for protecting Ni nanoparticles from agglomeration to improve durability even under reducing atmosphere.22 A drawback of organic capping reagents is the lack of the long-term stability under harsh reaction conditions. Additionally, the chemical interaction between organic molecules and catalyst surfaces may alter the surface properties of catalytic materials. Inorganic materials are durable enough to protect metal particles even under harsh reaction conditions with less interaction with metal surfaces. With inorganic materials, particles with core–shell structures, in which particles are covered with thermally and chemically stable porous materials, have been reported for catalytic application.37 For example, Pt nanoparticles covered with silica were effectively protected under high temperature conditions such as 973 K for alkane oxidation.38 A drawback of core–shell structures is the reduction of an available surface area of active core species. Even though the metal-oxide shell has a mesoporous structure, slow diffusion of a reactant reduces the apparent catalytic reactivity.
We report herein a novel protection method of Cu2O particles from agglomeration by partially covering with Co3O4 under highly reductive conditions. The Cu2O particles partially covered with Co3O4 nanoparticles showed the high catalytic reactivity for hydrogen evolution by AB hydrolysis with high durability. The catalytic reactivity and durability of the Cu2O–Co3O4 composites were compared with those of the Cu2O particles protected by conventional methods, i.e., capping with organic reagents or inorganic materials. As organic capping reagents, PVP, hexadecyltrimethylammonium bromide (CTAB) and sodium dodecyl sulfate (SDS) were used because of different electrical properties. As an inorganic capping and support material, mesoporous silica was used. Cu2O particles partially covered with Co3O4, where the Cu2O particles have the shape of 50-facets, cube, octahedron or rhombicuboctahedron, were also employed to clarify the shape effect of Cu2O particles on the catalytic reactivity for AB hydrolysis. Also the shape and size effects of Co3O4 nanoparticles were investigated on protection of Cu2O particles from agglomeration.
2. Experimental section
All chemicals used for synthesizing Cu2O or nanosized Co3O4 particles were obtained from a chemical company and used without further purification. Copper acetate monohydrate and copper(II) nitrate trihydrate were purchased from Nakalai Tesque and Wako Pure Chemical Industries, Ltd, respectively. Cobalt precursors of cobalt acetate tetrahydrate and cobalt nitrate hexahydrate were received from Kishida Chemicals and Wako Pure Chemical Industries, Ltd, respectively. Sodium dodecyl sulfate (SDS), sodium borohydride, D-(+)-glucose, ammonia solution (28%) and sodium hydroxide were obtained from Wako Pure Chemical Industries, Ltd. Poly-N-vinyl-2-pyrrolidone (PVP, K-15, Mw = 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000) and cetyltrimethylammonium bromide (CTAB) were obtained from Tokyo Chemical Industry. Ammonia borane, oleylamine, urea and 3-aminopropyltriethoxysilane (APTS) were received from Sigma-Aldrich Co. Tetraethoxysilane was received from Shin-Etsu Chemical Co., Ltd. Purified water was provided by a Millipore Milli-Q water purification system where the electronic conductance was 18.2 MΩ cm. Size and/or shape-controlled Cu2O or nanosized Co3O4 were synthesized by following reported methods.39–42
000) and cetyltrimethylammonium bromide (CTAB) were obtained from Tokyo Chemical Industry. Ammonia borane, oleylamine, urea and 3-aminopropyltriethoxysilane (APTS) were received from Sigma-Aldrich Co. Tetraethoxysilane was received from Shin-Etsu Chemical Co., Ltd. Purified water was provided by a Millipore Milli-Q water purification system where the electronic conductance was 18.2 MΩ cm. Size and/or shape-controlled Cu2O or nanosized Co3O4 were synthesized by following reported methods.39–42
2.1. Synthesis of particles
:0 (v/v) for the octahedron and 9:81 for the rhombicuboctahedron (RCO) and the cube] at 333 K. An aqueous solution (15 mL) of sodium hydroxide (5.0 M for cube and 10 M for RCO and octahedron) was dropped into the mixture with vigorous stirring at 60 °C. After 12 min stirring, an aqueous solution (45 mL) of D-(+)-glucose (0.9 g, 5.0 mmol) was slowly added to the suspension with vigorous stirring at 333 K for 60 min for the cube and 15 min for the RCO and the octahedron. The obtained particles were separated by centrifugation, washed with water three times and dried in vacuo for several hours.
2.2. Catalysts characterization
Transmittance electron microscope images of nanoparticles, which were mounted on a copper microgrid coated with elastic carbon, were observed by a JEOL JEM 2100 operating at 200 keV. Scanning electron microscope images of particles were observed by a FE-SEM (JSM-6320F or JSM-6701F) operating at 8.0 kV or 5.0 kV. Powder X-ray diffraction patterns were recorded by a Rigaku Ultima IV. Incident X-ray radiation was produced by a Cu X-ray tube, operating at 40 kV and 40 mA with CuKα radiation of 1.54 Å. The scanning rate was 2° min−1 from 20° to 80° in 2θ. X-Ray photon spectra were measured by a Kratos Axis 165× with a 165 mm hemispherical electron energy analyzer. The incident radiation was Mg Kα X-rays (1253.6 eV) at 200 W. Each sample was attached on a stainless stage with a double-sided carbon scotch tape. The binding energy of each element was corrected by the C 1s peak (248.6 eV) from residual carbon.2.3. Catalysis measurements
Catalytic particles (12 mg) were suspended in an aqueous solution of AB (25 mM, 20 mL) at 293 K in a glass tube (50 mL). The suspension was vigorously stirred with a magnetic stirrer during the reaction. The hydrogen evolved during the reaction was trapped in a gas burette on a water bath, and each volume was recorded with the reaction time.3. Results and discussion
3.1. Cu2O particles capped with organic reagents and inorganic materials
The catalytic reactivity of Cu2O particles capped with organic reagents was examined for hydrogen evolution from ammonia borane (AB) hydrolysis at 293 K. Prior to catalysis measurements, a capping reagent of D-(+)-glucose used for synthesis of Cu2O particles was exchanged to one of the water soluble capping reagents chosen from poly-N-vinyl-2-pyrrolidone (PVP), cetyltrimethylammonium bromide (CTAB) and sodium dodecyl sulfate (SDS), which are electrically neutral, cationic and anionic surfactants, respectively. The Cu2O particles were suspended in an aqueous solution of AB (0.5 mmol, 20 mL water) with vigorous stirring. Fig. 1a depicts the time course of hydrogen evolution by AB hydrolysis with the Cu2O particles capped with different organic molecules. Hydrogen evolution was observed with all the Cu2O particles, however, the yield of hydrogen was as low as 20% after the reaction time of 60 min. At the beginning of the reaction (within 1 min), fast hydrogen evolution, in which the reaction rate was 0.17 mmol s−1gcat−1, was observed in all cases. This clearly indicates that the Cu2O is very active for the reaction at the beginning, however, it is deactivated very soon after this period. Fig. 1b shows a typical SEM image of Cu2O particles with an organic capping reagent after AB hydrolysis. This image clearly indicates agglomeration of Cu2O particles after the reaction. Powder X-ray diffraction measurements of the Cu2O particles after AB hydrolysis indicated that Cu2O was completely reduced to Cu metal after the reaction (see Fig. S1 in the ESI†). The reduction of Cu2O surfaces to Cu metal may weaken an ionic interaction between the organic capping reagents and the surface of catalytic particles. Thus, these organic capping reagents cannot protect Cu2O particles effectively from agglomeration.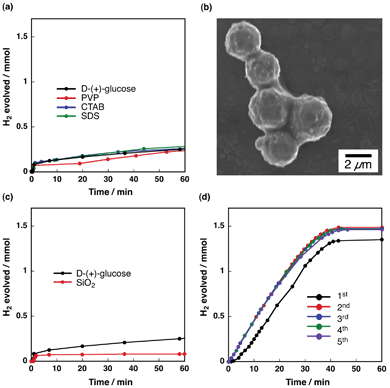 | ||
| Fig. 1 (a) Time course of hydrogen evolution with Cu2O(50-facets) with D-(+)-glucose (black), PVP (red), CTAB (blue) and SDS (green) as capping reagents. (b) A typical SEM image of Cu2O particles after AB hydrolysis. (c) Time course of hydrogen evolution with Cu2O covered with SiO2 shell. (d) Time course of hydrogen evolution with Cu2O loaded on SiO2 (SBA-15). The reaction was repeated 5 times under the same conditions. | ||
The catalytic reactivity of Cu2O particles covered with silica in a core–shell fashion (Cu2O@SiO2) was also examined for AB hydrolysis at 293 K. As indicated in Fig. 1c (red), a very small amount of hydrogen, which is less than 10% of the stoichiometric amount, was evolved. XPS spectra of Cu2O@SiO2 particles after the reaction shown in Fig. S2 (in the ESI†) indicate that silica coating is effective in avoiding agglomeration of Cu2O particles, however, most part of Cu2O-core surfaces could not be used for the catalytic reaction. When 5 wt% of Cu2O particles were loaded on a support of mesoporous silica (SBA-15), a stoichiometric amount of hydrogen was evolved within 50 min with the hydrogen evolution rate of 0.048 mmol s−1 gcat−1 as shown in Fig. 1d. The Cu2O/SBA-15 catalyst showed high stability without the loss of the reactivity for hydrogen evolution during 5 times repetitive use. These results clearly indicate that Cu2O particles act as catalysts for AB hydrolysis even after being contacted with SiO2. Thus, the very low reactivity of Cu2O@SiO2 results from less permeability of AB into pores of SiO2 shells.
3.2. Cu2O particles partially covered with Co3O4 nanoparticles
In order to maintain the surfaces of Cu2O particles accessible to AB, their surfaces were partially covered with metal oxide nanoparticles. Among metal-oxide nanoparticles active for AB hydrolysis, Co3O4 nanoplates can be expected to show the high catalytic reactivity by interacting with Cu2O.36 The Co3O4 plates were synthesized by the reported method and their sizes in between 100 nm and 500 nm and shapes of hexagonal plate were determined by TEM and SEM observations (vide infra, Fig. 7b). The calculated amount of Co3O4 plates and Cu2O particles were mixed and dispersed in water with sonication and heated to dryness at 353 K. Formation of Cu2O partially covered with Co3O4 (Cu2O–Co3O4) was confirmed by SEM, as shown in Fig. 2a (more SEM images of Cu2O–Co3O4 particles are displayed in Fig. S3 in the ESI†) with a schematic drawing of a Cu2O–Co3O4 composite in Fig. 2b. Catalytic hydrogen evolution by AB hydrolysis with Cu2O–Co3O4 (12 mg) was conducted in an aqueous solution of AB (0.50 mmol, 20 mL water) at 293 K. The time course of evolved hydrogen by AB hydrolysis with the Cu2O–Co3O4 is depicted in Fig. 2c (black). The time course of hydrogen evolution by AB hydrolysis with Co3O4 (blue) or Cu2O (red) is also shown as references. With Cu2O–Co3O4, hydrogen evolution was observed with a hydrogen-evolution rate of 0.31 mmol s−1 gcat−1 determined from the slope in between 1 and 4 min after starting the reaction. During the reaction, hydrogen of three times mol of AB should be evolved as given by eqn (1):| NH3–BH3 + 2H2O → NH4+ + BO2− + 3H2 | (1) |
![(a) SEM image of Cu2O(50-facets) decorated with Co3O4 nanoplates [Cu2O/Co3O4 = 1/1 (w/w)]. (b) Schematic drawing of decorated particles. (c) Time course of hydrogen evolution from AB hydrolysis with Cu2O decorated with Co3O4 nanoplates [black, 1 : 1(w/w)], Cu2O and Co3O4 nanoplates separately reacted in an H-type cell [green, 1 : 1(w/w)], Co3O4 nanoplates pretreated by H2 gas (blue) and Cu2O with d-(+)-glucose (red) [NH3BH3, 0.5 mmol; catalyst, 12 mg; water, 20 mL].](/image/article/2012/EE/c1ee02639a/c1ee02639a-f2.gif) | ||
| Fig. 2 (a) SEM image of Cu2O(50-facets) decorated with Co3O4 nanoplates [Cu2O/Co3O4 = 1/1 (w/w)]. (b) Schematic drawing of decorated particles. (c) Time course of hydrogen evolution from AB hydrolysis with Cu2O decorated with Co3O4 nanoplates [black, 1:1(w/w)], Cu2O and Co3O4 nanoplates separately reacted in an H-type cell [green, 1:1(w/w)], Co3O4 nanoplates pretreated by H2 gas (blue) and Cu2O with D-(+)-glucose (red) [NH3BH3, 0.5 mmol; catalyst, 12 mg; water, 20 mL]. | ||
Thus, the stoichiometric amount of hydrogen evolved in this system is 1.5 mmol. When the Cu2O–Co3O4 was used as a catalyst, the volume of hydrogen evolved was 1.3 mmol, which corresponds to 87% of the stoichiometric amount of hydrogen. When Co3O4 alone was used as a catalyst, a long induction period, such as 60 min, was required to show the catalytic reactivity without a reductive pretreatment.32,36 Even when Co3O4 was pretreated with hydrogen gas under the reaction conditions, no hydrogen evolution was observed in 10 min (Fig. 2c, blue). The catalytic reactivity was also examined for the reaction system with an H-type cell where Co3O4 and Cu2O were separately put in two tubes of the H-type cell to evaluate the effect of interaction between Co3O4 and Cu2O. As shown in Fig. 2c (green), only a small amount of hydrogen was evolved from the tube containing Cu2O. Thus, the interaction between Cu2O and Co3O4 is indispensable to achieve the high catalytic reactivity.
The repetitive catalytic tests with Cu2O–Co3O4 showed no significant reactivity loss for three times use as shown in Fig. 3a. The hydrogen-evolution rates of the 2nd and 3rd cycles were 0.31 and 0.32 mmol s−1gcat−1, respectively. For the 1st cycle, a short induction period of 1 min was required before continuous hydrogen evolution, however, the induction period observed for the 1st cycle was not observed for the 2nd and 3rd cycles. Also the amount of hydrogen evolved for the 2nd and 3rd cycles reached 1.45 mmol, which corresponds to 96% of the stoichiometric amount. These results indicate that a small amount of hydrogen evolved in the induction period was consumed for reducing the catalyst surfaces in the 1st cycle. SEM observations of Cu2O–Co3O4 particles after the reaction also assure no agglomeration of Cu2O particles as shown in Fig. 3b. An SEM image of high magnification depicts the presence of Co3O4 particles on Cu2O surfaces. The presence of Co3O4 nanoparticles effectively prevents the agglomeration of Cu2O particles to improve durability of the catalysis.
![(a) Repetitive test (3 times) of hydrogen evolution by AB hydrolysis with Cu2O–Co3O4 [Cu2O/Co3O4 = 1/1 (w/w)]. (b) SEM image of Cu2O(50-facets) decorated with Co3O4 nanoplates after reaction (NH3BH3, 0.5 mmol; catalyst, 12 mg; water, 20 mL).](/image/article/2012/EE/c1ee02639a/c1ee02639a-f3.gif) | ||
| Fig. 3 (a) Repetitive test (3 times) of hydrogen evolution by AB hydrolysis with Cu2O–Co3O4 [Cu2O/Co3O4 = 1/1 (w/w)]. (b) SEM image of Cu2O(50-facets) decorated with Co3O4 nanoplates after reaction (NH3BH3, 0.5 mmol; catalyst, 12 mg; water, 20 mL). | ||
The surface conditions of the Cu2O–Co3O4 composite before and after AB hydrolysis were examined by X-ray photoelectron spectroscopy (XPS) as shown in Fig. 4. The measurements were performed for the binding energy fields of Cu 2p, Co 2p and O 1s. As shown in Fig. 4a, Cu 2p peaks of the fresh catalyst were complicated, because a part of the Cu(I) species were oxidized to Cu(II) during the composite preparation. The satellite peaks characteristic of Cu(II) species diminished in the Cu 2p peaks of the used catalyst (Fig. 4d), indicating low valent Cu species formed after the reaction.36 For the Co 2p peaks of used catalysts (Fig. 4e), the strong satellite peaks were observed at 802.5 eV and 786.2 eV, which were very weakly observed in the fresh catalyst. These strong satellite peaks clearly indicated the presence of Co(II) species in the used catalyst.36 Also, the O 1s peak of the used catalyst appeared at 531.2 eV (Fig. 4f), which was shifted from 530.0 eV of the fresh catalyst (Fig. 4c) corresponding to the formation of –OH species on the surface of used catalyst.36 Thus, both Cu2O and Co3O4 species have been reduced and formed corresponding hydroxide species during the reaction.
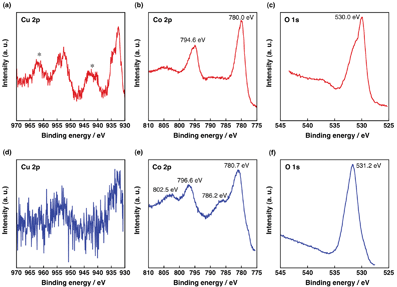 | ||
| Fig. 4 X-Ray photoelectron spectra of the fresh (a–c) and used (d–f) catalysts of the Cu2O–Co3O4 composite in the binding energy regions of Cu 2p (a and d), Co 2p (b and e) and O 1s (c and f) (* denotes satellite peak). | ||
The catalytic reactivity of Cu2O–Co3O4 for AB hydrolysis was examined with different Cu2O/Co3O4 ratios, 5/1 and 1/5 (w/w), under the same conditions for the Cu2O–Co3O4 (1/1) catalyst. The catalytic hydrogen evolution by AB hydrolysis was conducted in an aqueous solution containing AB (0.5 mmol) with a catalyst (12 mg). When the catalyst contained a smaller amount of Co3O4 with a Cu2O/Co3O4 ratio of 5/1 (5Cu2O–Co3O4), a smaller amount of hydrogen of 1.1 mmol was evolved than Cu2O–Co3O4 and the hydrogen-evolution rate was 0.13 mmol s−1gcat−1, which is less than half of the hydrogen-evolution rate observed for Cu2O–Co3O4 as shown in Fig. 5. When the catalyst with a Co3O4-rich composition of Cu2O/Co3O4 = 1/5 (Cu2O–5Co3O4) was employed, the hydrogen-evolution rate was slightly increased to 0.38 mmol s−1 gcat−1. The successful suppression of Cu2O-particles agglomeration by partial coverage with Co3O4 nanoparticles encouraged us to examine the effect of Cu2O shapes on the catalytic reactivity.
![Time course of hydrogen evolution by AB hydrolysis with Cu2O/Co3O4 catalysts with different Cu2O/Co3O4 ratios [circle, 1/1 (w/w); square, 5/1; triangle, 1/5] (NH3BH3 = 0.5 mmol, catalyst = 12 mg, water = 20 mL).](/image/article/2012/EE/c1ee02639a/c1ee02639a-f5.gif) | ||
| Fig. 5 Time course of hydrogen evolution by AB hydrolysis with Cu2O/Co3O4 catalysts with different Cu2O/Co3O4 ratios [circle, 1/1 (w/w); square, 5/1; triangle, 1/5] (NH3BH3 = 0.5 mmol, catalyst = 12 mg, water = 20 mL). | ||
3.3. Effect of Cu2O shape on catalysis for ammonia borane hydrolysis
Fig. 6 shows SEM images of Cu2O particles with different shapes prepared by the reported method.39 The particles were prepared with D-(+)-glucose as a capping reagent. The size of Cu2O with the shape of 50-facets [Cu2O(50-facets)] is nearly 2 μm (Fig. 6a). As indicated in Fig. 6b, the size of cube-shaped Cu2O [Cu2O(cube)] was 500 nm (vertex–vertex). The Cu2O particles with octahedron shape [Cu2O(octahedron)] have 0.5–0.7 μm side length. The Cu2O particles with rhombicuboctahedron shape [Cu2O(RCO)] showed 0.8–1 μm. The crystal phase of these Cu2O particles was determined by powder X-ray diffraction patterns. The diffraction peaks at 2θ = 29.6°, 36.5°, 42.3°, 61.4°, 73.6° and 77.4° are indexed to (110), (111), (200), (220), (311) and (222) planes, respectively, by comparison with reported values.39 | ||
| Fig. 6 SEM images of Cu2O particles with the shape of (a) 50-facets, (b) cube, (c) octahedron and (d) rhombicuboctahedron. A typical powder X-ray diffraction pattern achieved for Cu2O particles. The numbers in parenthesis are (hkl) indexes. | ||
Cu2O particles with different shapes of 50-facets, cube, octahedron and RCO were decorated with Co3O4 rods (vide infra)45 with the ratio of Cu2O/Co3O4 = 1/5. The hydrogen-evolution rates normalized by weights of Cu2O–Co3O4 or surface areas of Cu2O are summarized in Table 1. Specific surface areas were calculated from the average sizes and distributions of nanoparticles determined by SEM measurements. The fastest hydrogen-evolution rate normalized by catalyst weight was obtained for Cu2O(cube)–5Co3O4 as 0.78 mmol s−1 gcat−1. The hydrogen-evolution rate for Cu2O(RCO)–5Co3O4 was 0.67 mmol s−1gcat−1, which is higher than the rate for Cu2O(50-facets)–5Co3O4 of 0.56 mmol s−1 gcat−1. On the other hand, the hydrogen-evolution rate normalized per surface area of Cu2O was the largest at Cu2O(50-facets)–5Co3O4 as 9.1 mmol s−1mCu2O−2, which is more than two times larger than the rate of 3.6 mmol s−1mCu2O−2 with Cu2O(RCO)–5Co3O4. The surface of Cu2O(50-facets) consists of (100), (110), (111) and (311) planes. Each plane occupies 14.7%, 39.1%, 11.3% and 34.9% of the total surface area, respectively. The surface of Cu2O(RCO) is composed of (100), (110) and (111) surfaces, where the (311) plane is excluded. Also, the surfaces of Cu2O(cube) and Cu2O(octahedron) are exclusively composed of the (100) surface and the (111) surface, respectively. Thus, the high catalytic reactivity of Cu2O(50-facets) normalized by the surface area of Cu2O resulted from the (311) plane exclusively included in the surface of the Cu2O(50-facets). Such high reactivity of the (311) plane of Cu2O has been reported for CO oxidation39 and predicted from theoretical calculations.44
| Shape | Hydrogen-evolution rate | Cu2O surface area/m2 g−1 | |
|---|---|---|---|
| mmol s−1gcat−1 | mmol s−1mCu2O−2 | ||
| a Reaction conditions [Cu2O/Co3O4 = 1/5 (w/w); NH3BH3, 0.5 mmol; catalyst, 12 mg; water, 20 mL] (time course of hydrogen evolution is displayed in Fig. S4 in the ESI†). b Rhombicuboctahedron. | |||
| 50-Facets | 0.56 | 9.1 | 0.37 |
| Cube | 0.78 | 2.1 | 2.2 |
| Octahedron | 0.44 | 1.1 | 2.3 |
| RCO b | 0.67 | 3.6 | 1.1 |
3.4. Effect of Co3O4 shape on catalysis for NH3–BH3 hydrolysis
The shape of Co3O4 nanoparticles can influence the catalytic reactivity of Cu2O–Co3O4 composites by changing interaction between Cu2O and Co3O4. Co3O4 nanoparticles in the shape of a sphere, a hexagonal plate and a porous rod were used for decorating Cu2O(50-facets). These shape-controlled Co3O4 nanoparticles were synthesized by reported methods40–42 and their sizes and shapes were determined by TEM observation as shown in Fig. 7a–c. As shown in Fig. 7a, the sizes of spherical particles of Co3O4 [Co3O4(sphere)] are in the range between 2 and 6 nm with an average size of 4 nm. Co3O4 nanoplates [Co3O4(plate)] have hexagonal shape and the sizes of the particles are in the range between 500 nm and 1 μm (vertex to vertex) with the thickness of 10–25 nm (Fig. 7b). Co3O4 nanoparticles with rod shape [Co3O4(rod)] have a porous structure and the size is 50–250 nm in width and 0.5–10 μm in length Fig. 7c. | ||
| Fig. 7 TEM images of Co3O4 nanoparticles with the shape of (a) sphere, (b) nanoplate and (c) rod. (d) SEM image of Cu2O(50-facets)–Co3O4(rod). | ||
Calculated amounts of the size- and/or shape-controlled Co3O4 nanoparticles and Cu2O particles with the shape of 50-facets were mixed in water following sonication and dried at 353 K. The weight ratios of Cu2O/Co3O4 were selected from 1/5, 1/1 and 5/1. A typical SEM image of a composite of Cu2O(50-facets)–Co3O4(rod) is displayed in Fig. 7d. The image clearly indicates that the diameter of the rod as well as the thickness of the nanoplate (Fig. S3 in the ESI†) are critical parameters for the separation of Cu2O particles. No clear SEM image was obtained for Cu2O(50-facets)–Co3O4(sphere) because Co3O4(sphere) is too small (2–6 nm) to be observed by SEM. Fig. 8 indicates the time courses of hydrogen evolution with Cu2O–Co3O4 composites. The catalytic tests were repeated three times for each catalyst. Fig. 8a indicates the time course of hydrogen evolution by AB (0.5 mmol) hydrolysis with 5Cu2O–Co3O4 (12 mg) where the shapes of Co3O4 nanoparticles were sphere (black), nanoplate (red) or rod (blue). The volume of evolved hydrogen reached 1.44 mmol for the 2nd and 3rd cycles, which is 96% of the stoichiometric amount, when 5Cu2O(50-facets)–Co3O4(plate) and 5Cu2O(50-facets)–Co3O4(rod) were used as catalysts. The slightly lower yield of the 1st cycle is due to the use of formed hydrogen for the reduction of surface of Cu2O–Co3O4 composites. On the other hand, when 5Cu2O(50-facets)–Co3O4(sphere) was used as a catalyst, the amount of evolved hydrogen was 1.2, 1.3 and 1.2 mmol for the 1st, 2nd and 3rd cycle, respectively. The lower hydrogen yields, especially at the 3rd cycle, indicate that Cu2O(50-facets) starts to agglomerate because Co3O4(sphere) is too small to protect large Cu2O particles. The large Co3O4(plate) and Co3O4(rod) particles effectively separate each Cu2O particle not to form agglomerates, leading to high hydrogen yields even at the 2nd and 3rd cycles. When the ratio of Co3O4 increased to Cu2O/Co3O4 = 1/5, the catalytic reactivity of Cu2O(50-facets)–5Co3O4(sphere) was improved to the same level of Cu2O(50-facets)–5Co3O4(plate) and Cu2O(50-facets)–5Co3O4(rod) in terms of hydrogen-evolution rates and hydrogen yields as shown in Fig. 8b. By increasing the relative amount of Co3O4, even smaller particles of Co3O4(sphere) can protect Cu2O particles from agglomeration.
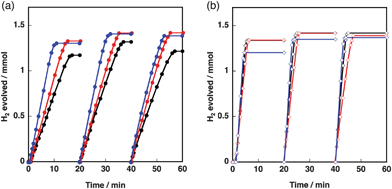 | ||
| Fig. 8 Time course of hydrogen evolution by AB hydrolysis with Cu2O particles (50-facets) partially covered with shape-controlled Co3O4 (black, sphere; red, plate; blue, rod) at room temperature (NH3BH3, 0.5 mmol; catalyst, 12 mg; water, 20 mL). The ratios of Cu2O/Co3O4 (w/w) were (a) 5/1 and (b) 1/5. The data for Cu2O/Co3O4 (1/1) are shown in Fig. S5 in the ESI†. | ||
The reactivity data shown in Fig. 8 provided a clue to discuss active species where the same Cu2O particles were used for the preparation of Cu2O–Co3O4 composites. Co3O4(sphere) is as small as 4 nm, whereas Co3O4(plate) and Co3O4(rod) are much larger (see Fig. 2a and 7). If the actual active species for the reaction were Co species supported on Cu2O, Cu2O with smaller Co3O4(sphere) should have shown much higher reactivity than Cu2O with larger Co3O4(plate or rod), because smaller Co3O4 particles have higher surface area and can more readily interact with Cu2O particles. However, as indicated in Fig. 8a, 5Cu2O–Co3O4(sphere, black) showed slower H2 evolution rate than 5Cu2O–Co3O4(rod, blue, and nanoplate, red). Additionally, as shown in Fig. 8b and S5†, Cu2O–Co3O4 composites containing a higher amount of Co3O4 showed similar reactivity without Co3O4 shape dependence, which was observed for Co3O4 catalysts in AB hydrolysis with NaBH4.36 From Table 1 and Fig. S4†, the catalytic reactivity of Cu2O–5Co3O4 composites depends on the shape of Cu2O. Thus, Cu2O particles play an important role to determine the catalytic performance of current Cu2O–Co3O4 composites under the current reaction conditions although Co species may have a certain contribution to the catalytic activity of Cu2O–Co3O4 composites.
The catalytic reactivity examined for a physical mixture of Cu2O(50-facets) and Co3O4(rod) was similar to that of Cu2O(50-facets)–Co3O4(rod) composites. This indicates that the particles of Cu2O and Co3O4 form composites without any special operations under reaction conditions. Under neutral conditions, Cu2O weakly interacted with Co3O4 by electrostatic interactions. Alternatively under reaction conditions, the interaction among Cu2O particles and Co3O4 nanoparticles becomes stronger because some of the surface oxygen atoms of Cu2O are reducibly removed to form oxygen vacant sites, which can interact with the surface oxygen of Co3O4 or Co(OH)2 species.
Conclusions
Agglomeration of Cu2O particles under reduced conditions was effectively suppressed by the partial coverage with Co3O4 nanoparticles to exhibit the high catalytic reactivity for hydrogen evolution by ammonia borane (AB) hydrolysis. The novel method to protect Cu2O particles from agglomeration enables us to examine the shape effect of Cu2O particles on their catalysis in AB hydrolysis under highly reducing conditions for the first time. Among four Cu2O particles with the shape of 50-facets, cube, octahedron and rhombicuboctahedron, Cu2O with the shape of 50-facets showed the highest specific reactivity per surface area. This may be ascribed to the higher surface energy of a high-index plane of (311) than those of low index surfaces. Additionally, the conditions to use small sized Co3O4 nanoparticles for protecting Cu2O particles were clarified. When small Co3O4 particles were used for protecting large Cu2O particles, a high Co3O4 ratio in the Cu2O/Co3O4 composite was required to achieve sufficient effect for protecting Cu2O particles from agglomeration. The method to protect catalytic particles with smaller nanoparticles disclosed here for the first time provides an opportunity for metal particles, which easily agglomerate under harsh reaction conditions, to be utilized as durable catalysts.Acknowledgements
This work was supported by a Grant-in-Aid (no. 20108010), and a Global COE program, “the Global Education and Research Centre for Bio-Environmental Chemistry” from the Ministry of Education, Culture, Sports, Science and Technology (MEXT), Japan (to S.F.), NRF/MEST of Korea through WCU (R31-2008-000-10010-0) and GRL (2010-00353) Programs (to S.F.). Y.Y. acknowledges the financial support from the Ogasawara Foundation for the Promotion of Science and Engineering. We sincerely acknowledge the Research Center for Ultra-Precision Science & Technology for TEM measurements.Notes and references
- A. Staubitz, A. P. M. Robertson and I. Manners, Chem. Rev., 2010, 110, 4079–4124 CrossRef CAS.
- B. Peng and J. Chen, Energy Environ. Sci., 2008, 1, 479–483 CAS.
- Q. Xu and M. Chandra, J. Alloys Compd., 2007, 446–447, 729–732 CrossRef CAS.
- D. G. Nocera, Chem. Soc. Rev., 2009, 38, 13–15 RSC.
- S. Fukuzumi, Eur. J. Inorg. Chem., 2008, 1351–1362 CrossRef CAS.
- H. L. Jiang, S. K. Singh, J. M. Yan, X. B. Zhang and Q. Xu, ChemSusChem, 2010, 3, 541–549 CrossRef CAS.
- T. Umegaki, J.-M. Yan, X.-B. Zhang, H. Shioyama, N. Kuriyama and Q. Xu, Int. J. Hydrogen Energy, 2009, 34, 2303–2311 CrossRef CAS.
- M. Chandra and Q. Xu, J. Power Sources, 2006, 156, 190–194 CrossRef CAS.
- T. M. Douglas, A. B. Chaplin, A. S. Weller, X. Yang and M. B. Hall, J. Am. Chem. Soc., 2009, 131, 15440–15456 CrossRef CAS.
- D. W. Himmelberger, C. W. Yoon, M. E. Bluhm, P. J. Carroll and L. G. Sneddon, J. Am. Chem. Soc., 2009, 131, 14101–14110 CrossRef CAS.
- M. Chandra and Q. Xu, J. Power Sources, 2006, 159, 855–860 CrossRef CAS.
- H. L. Jiang and Q. Xu, Catal. Today, 2011, 170, 56–63 CrossRef CAS.
- F. Y. Cheng, H. Ma, Y. M. Li and J. Chen, Inorg. Chem., 2007, 46, 788–794 CrossRef CAS.
- N. Mohajeri, A. T. Raissi and O. Adebiyi, J. Power Sources, 2007, 167, 482–485 CrossRef CAS.
- J. H. Park, H. S. Kim, H. J. Kim, M. K. Han and Y. G. Shul, Res. Chem. Intermed., 2008, 34, 709–715 CrossRef CAS.
- S. Basu, A. Brockman, P. Gagare, Y. Zheng, P. V. Ramachandran, W. N. Delgass and J. P. Gore, J. Power Sources, 2009, 188, 238–243 CrossRef CAS.
- F. Durap, M. Zahmakiran and S. Ozkar, Int. J. Hydrogen Energy, 2009, 34, 7223–7230 CrossRef CAS.
- S. B. Kalidindi, J. Joseph and B. R. Jagirdar, Energy Environ. Sci., 2009, 2, 1274–1276 CAS.
- S. B. Kalidindi, A. A. Vernekar and B. R. Jagirdar, Phys. Chem. Chem. Phys., 2009, 11, 770–775 RSC.
- Y. Q. Li, L. Xie, Y. Li, J. Zheng and X. G. Li, Chem.–Eur. J., 2009, 15, 8951–8954 CrossRef CAS.
- O. Metin, S. Sahin and S. Ozkar, Int. J. Hydrogen Energy, 2009, 34, 6304–6313 CrossRef CAS.
- T. Umegaki, J. M. Yan, X. B. Zhang, H. Shioyama, N. Kuriyama and Q. Xu, Int. J. Hydrogen Energy, 2009, 34, 3816–3822 CrossRef CAS.
- T. Umegaki, J. M. Yan, X. B. Zhang, H. Shioyama, N. Kuriyama and Q. Xu, J. Power Sources, 2009, 191, 209–216 CrossRef CAS.
- J. M. Yan, X. B. Zhang, S. Han, H. Shioyama and Q. Xu, Inorg. Chem., 2009, 48, 7389–7393 CrossRef CAS.
- J. M. Yan, X. B. Zhang, S. Han, H. Shioyama and Q. Xu, J. Power Sources, 2009, 194, 478–481 CrossRef CAS.
- J. M. Yan, X. B. Zhang, H. Shioyama and Q. Xu, J. Power Sources, 2010, 195, 1091–1094 CrossRef CAS.
- X. J. Yang, F. Y. Cheng, J. Liang, Z. L. Tao and J. Chen, Int. J. Hydrogen Energy, 2009, 34, 8785–8791 CrossRef CAS.
- F. Durap, M. Zahmakiran and S. Ozkar, Appl. Catal., A, 2009, 369, 53–59 CrossRef CAS.
- M. Zahmakiran and S. Ozkar, Appl. Catal., B, 2009, 89, 104–110 CrossRef CAS.
- V. I. Simagina, P. A. Storozhenko, O. V. Netskina, O. V. Komova, G. V. Odegova, Y. V. Larichev, A. V. Ishchenko and A. M. Ozerova, Catal. Today, 2008, 138, 253–259 CrossRef CAS.
- O. Metin and S. Ozkar, Energy Fuels, 2009, 23, 3517–3526 CrossRef CAS.
- S. B. Kalidindi, M. Indirani and B. R. Jagirdar, Inorg. Chem., 2008, 47, 7424–7429 CrossRef CAS.
- T. J. Clark, G. R. Whittell and I. Manners, Inorg. Chem., 2007, 46, 7522–7527 CrossRef CAS.
- T. He, Z. T. Xiong, G. T. Wu, H. L. Chu, C. Z. Wu, T. Zhang and P. Chen, Chem. Mater., 2009, 21, 2315–2318 CrossRef CAS.
- S. B. Kalidindi, U. Sanyal and B. R. Jagirdar, Phys. Chem. Chem. Phys., 2008, 10, 5870–5874 RSC.
- Y. Yamada, K. Yano, Q. Xu and S. Fukuzumi, J. Phys. Chem. C, 2010, 114, 16456–16462 CAS.
- A. Guerrero-Martinez, J. Perez-Juste and L. M. Liz-Marzan, Adv. Mater., 2010, 22, 1182–1195 CrossRef CAS.
- S. H. Joo, J. Y. Park, C.-K. Tsung, Y. Yamada, P. Yang and G. A. Somorjai, Nat. Mater., 2008, 8, 126–131 CrossRef.
- M. Leng, M. Liu, Y. Zhang, Z. Wang, C. Yu, X. Yang, H. Zhang and C. Wang, J. Am. Chem. Soc., 2010, 132, 17084–17087 CrossRef CAS.
- Y. M. Dong, K. He, L. Yin and A. M. Zhang, Nanotechnology, 2007, 18, 6 Search PubMed.
- L. H. Hu, Q. Peng and Y. D. Li, J. Am. Chem. Soc., 2008, 130, 16136–16137 CrossRef CAS.
- Y. Zhang, Y. Chen, J. Zhou, T. Wang and Y. Zhao, Solid State Commun., 2009, 149, 585–588 CrossRef CAS.
- C.-T. Tu, A.-Q. Wang, M.-Y. Zheng, X.-D. Wang and T. Zhang, Appl. Catal., A, 2006, 297, 40–47 CrossRef CAS.
- A. Soon, X.-Y. Cui, B. Delley, S.-H. Wei and C. Stampfl, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 79, 035205–035215 CrossRef.
- Co3O4 with the rod shape was used for shape-controlled Cu2O particles protection because the catalytic reactivity of Cu2O–Co3O4 (rod) was slightly higher than Cu2O–Co3O4 (plate) as shown in Fig. 8.
Footnote |
| † Electronic supplementary information (ESI) available: Powder XRD pattern after the reaction (Fig. S1), XPS data of Cu2O@SiO2 (Fig. S2), SEM images of Cu2O–Co3O4 composites (Fig. S3) and time course of hydrogen evolution (Fig. S4 and S5). See DOI: 10.1039/c1ee02639a |
| This journal is © The Royal Society of Chemistry 2012 |