The diazofluorene antitumor antibiotics: Structural elucidation, biosynthetic, synthetic, and chemical biological studies
Seth B.
Herzon
* and
Christina M.
Woo
Yale University – Chemistry, 225 Prospect Street, P.O. Box 208107, New Haven, Connecticut 06520-8107, United States
First published on 28th October 2011
Abstract
Covering: up to 2011
This review presents a comprehensive survey of all aspects of the kinamycins and lomaiviticins, potent antiproliferative antimicrobial metabolites isolated from various strains of Streptomyces and Salinispora. The kinamycins and lomaiviticins contain a diazotetrahydrobenzo[b]fluorene (diazofluorene) functional group, which is unique among known natural products. This review begins with an account of the studies leading to the final (correct) structure determination of the kinamycins, which were originally proposed to contain an N-cyano carbazole function. This is followed by a discussion of biosynthetic studies, which established the polyketide nature of the kinamycins. Descriptions of four completed syntheses of various kinamycins, synthetic studies toward the lomaiviticins, syntheses of the carbohydrates of the lomaiviticins, and syntheses of structurally-related metabolites, are then presented. A survey of chemical biological investigations, including in vitro reactivity studies, which indicate that the kinamycins and lomaiviticins may form reactive ortho-quinone methide or free radical intermediates in vivo, is presented. Finally, a selection of structurally-related metabolites are described.
 Seth B. Herzon | Seth B. Herzon was born in Philadelphia, Pennsylvania in 1979. He received his BS in Chemistry from Temple University (2002) and his PhD from Harvard University (2006), under the guidance of Professor Andrew G. Myers. From 2006–2008 he was a postdoctoral fellow in the laboratory of Professor John F. Hartwig at the University of Illinois, Urbana–Champaign. In 2008, he began his independent career at Yale University. His research focuses on natural products synthesis. |
 Christina M. Woo | Christina Woo was born in Syracuse, New York in 1986. She received her BA in Chemistry from Wellesley College (2008). In 2008, she began graduate studies at Yale University in the laboratory of Professor Seth Herzon. Her research focuses on synthetic and chemical biological studies of the kinamycins, lomaiviticins, and unnatural diazofluorene-based anticancer agents. |
1 Introduction and isolation
The kinamycins and lomaiviticins are complex bacterial metabolites that were isolated from various strains of Streptomyces and Salinispora.1 Owing to the presence of a common diazotetrahydrobenzo[b]fluorene substructure, these metabolites are often referred to as “diazofluorenes” (vide infra). Kinamycins A–D (1–4, Table 1) are the earliest known members of this family. They were isolated by Ōmura and co-workers as yellow or orange needles from a strain of Streptomyces murayamaensis, itself obtained from a soil sample in Murayama, Saitama-ken, Japan.2–5 Subsequently, many different kinamycins have been isolated, and these are differentiated primarily by the acylation pattern about the D-ring (Table 1).6–11 All of the metabolites in Table 1 are levorotatory, with the exception of kinamycins E (5) and I (9), whose optical rotation does not appear to have been determined.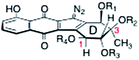
The related dimeric metabolites lomaiviticins A and B (11 and 12) were isolated by He and co-workers in 2001 (Fig. 1).12 The lomaiviticins bear some structural homology to the kinamycins, but important differences exist between the two classes of natural products. Most notably, the lomaiviticins possess two monomeric diazofluorenes united by a carbon–carbon bond [and in the case of lomaiviticin B (12), two carbon–oxygen bonds], to form a C2-symmetric structure. The naphthoquinone substructure of the lomaiviticins incorporates two hydroxyl groups (rather than a single hydroxyl group, as in the kinamycins), the tertiary alcohol functions (C-3) of the lomaiviticins possess an ethyl substituent (rather than methyl, as in the kinamycins), the C-1 position of the lomaiviticins are at the carbonyl oxidation state (rather than the alcohol oxidation state, as in the kinamycins), and the periphery of the lomaiviticins are functionalized with 2–4 2,6-dideoxyglycoside residues, oleandrose and N,N-dimethylpyrrolosamine (rather than acyl substituents, as in the kinamycins).
![Structures of lomaiviticin A (11), lomaiviticin B (12), and diazotetrahydrobenzo[b]fluorene (13).](/image/article/2012/NP/c1np00052g/c1np00052g-f1.gif) | ||
| Fig. 1 Structures of lomaiviticin A (11), lomaiviticin B (12), and diazotetrahydrobenzo[b]fluorene (13). | ||
The relationship between lomaiviticins A (11) and B (12) is, at present, not known. Given the hydrolytic instability of 2-deoxyglycosides,13 it is conceivable that lomaiviticin B (12) may arise from lomaiviticin A (11) by hydrolysis of the oleandrose residues (see structure 14) followed by cyclization (Scheme 1). Alternatively, lomaiviticin B (12) may be a biosynthetic precursor to lomaiviticin A (11) by direct glycosylation, or the two may diverge from a common intermediate earlier in the biosynthetic pathway.
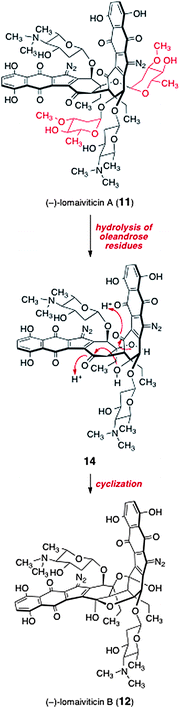 | ||
| Scheme 1 Potential mechanism for the conversion of lomaiviticin A (11) to lomaiviticin B (12). | ||
Structurally, the kinamycins and lomaiviticins contain a diazocyclopentadiene fused to an oxygenated naphthoquinone and a partially unsaturated six-membered ring. This substructure constitutes a diazotetrahydrobenzo[b]fluorene (see structure 13, Fig. 1), and these metabolites are often referred to as diazofluorenes. Because the diazofluorene macrofunctional group was unprecedented in natural products, the structural elucidation of the kinamycins was not straightforward (vide infra). The extended π-conjugation of the diazofluorene, coupled with the peripheral oxygen substituents, makes it difficult to predict the reactivity of the constituent functionality. From a synthetic standpoint, this generates many exciting questions relating to stability, reagent compatibility, and strategy.
The kinamycins and lomaiviticins are potent anticancer agents, with 50 percent inhibitory potencies at nanomolar–picomolar concentrations. The activity profiles of kinamycin C (3)14 and lomaiviticin A (11)12 are unique, suggesting a novel mechanism of action is operative. Both the kinamycins and lomaviticins have shown activity against Gram-positive and Gram-negative bacteria.12
Reviews of the kinamycins,15,16 diazo-containing natural products,17 and diazo-based anticancer agents18 have been previously published. This review seeks to cover all aspects of the chemistry and biology of the diazofluorene antitumor antibiotics, including the lomaiviticins, with an emphasis on synthetic and chemical biological research conducted since 2000.
2 Structure elucidation
2.1 The kinamycins
The structure elucidation of the kinamycins was a formidable challenge, and the pathway from isolation to structure determination involved the work of several research groups over a period of more than twenty years. The structural elucidation was challenging, in part, because at the time the kinamycins were isolated, high field NMR instruments and proton-detected multidimensional pulse sequences were not widely available. Moreover, the D-ring and juglone fragments of the kinamycins are separated by a large number of quaternary carbon atoms, preventing correlation of these spin systems by 3JH couplings. Perhaps most significantly, closely related compounds, which would be useful as benchmarks for spectroscopic comparison, were not well-known. As will be shown, the originally proposed structure for the kinamycins contained a cyanamide rather than a diazo function. Subsequent investigations led to permutation of the cyanamide atoms to a diazo function. This pathway from isolation to determination of the correct structure is described below.Initial spectroscopic and degradation studies of the kinamycins were conducted by Ōmura and co-workers in the early 1970s.2–5 These investigations established the constitution and relative stereochemistry of the D-ring and the juglone fragments of the kinamycins. Based on MS and IR analysis (strong absorption at ∼2150 cm−1), and the results of alkaline degradation (liberation of ammonia), a cyanamide function was suggested to bridge the juglone and D-ring substructures, leading to a unique N-cyanocarbazole function (15, Fig. 2). Shortly thereafter, an X-ray analysis of a para-bromobenzoate derivative of kinamycin C (3) was obtained, which established the absolute stereochemistry.19 However, the quality of the X-ray data did not permit rigorous assignment of the putative cyanamide function. Instead, the presence of this function was inferred based on Ōmura's earlier studies. It was noted20 that the carbon-13 resonance of the putative cyanamide carbon of kinamycin D (4) was not observed. This was attributed to an unusually long relaxation time or obfuscation by other signals.
![Kinamycin D [15, original (incorrect) structure], N-cyanoindoloquinone 16, and prekinamycin [17, original (incorrect) structure].](/image/article/2012/NP/c1np00052g/c1np00052g-f2.gif) | ||
| Fig. 2 Kinamycin D [15, original (incorrect) structure], N-cyanoindoloquinone 16, and prekinamycin [17, original (incorrect) structure]. | ||
In the late 1980s and early 1990s, the results of several studies began to generate uncertainty surrounding the N-cyanocarbazole. In 1988, Gould demonstrated that fermentation of Streptomyces murayamaensis in the presence of (15NH4)2SO4 produced [15N2]-kinamycin D.21 The putative cyanamide carbon was unequivocally identified by distinct C, N couplings in the proton-decoupled carbon-13 NMR spectrum. This carbon resonated at δ 78.5 (dd, JC,N = 21.2, 5.4 Hz). Careful re-inspection of 13C NMR data of natural kinamycin D (4) revealed a small singlet at the same resonance, nearly obscured by the chloroform-d signal. Gould noted that this chemical shift was ca. 30 ppm upfield of typical cyanamide resonances.
In 1990, Dmitrienko and co-workers reported the synthesis and characterization of a series of N-cyanoindoloquinones, as exemplified by structure 16 (Fig. 2).22 These workers observed that the resonance of the cyanamide carbon was in the δ ∼ 105 range, as expected based on literature precedent. Additionally, the cyanamide infrared stretch of 16 was in the 2237–2259 cm−1 range, vs. 2119–2170 cm−1 for the kinamycins.
In 1993, Echavarren and co-workers reported the preparation of structure 17,23 which was referred to as prekinamycin (itself isolated6,8 from the producing strain of the kinamycins). The cyanamide carbon of Echavarren's synthetic material (17) was observed at δ ∼ 112, clearly distinct from the shift reported for natural prekinamycin (δ 83.7).81H spectroscopic data of synthetic and natural prekinamycin were also incongruent. Collectively, these studies called into question the original structural assignment of prekinamycin and the kinamycins.
The matter was finally settled in 1994 in back-to-back communications by Gould24 and Dmitrienko.25 Gould presented two critical data points. First, he showed that treatment of natural prekinamycin with dirhodium tetracetate in methanol yielded the fluorene 19 (Scheme 2). The location of the newly formed vinyl proton (H-1, δ 7.25) was established by observation of an NOE to H-2 and a 3JC,H coupling to the semiquinone phenolic carbon. In parallel, his research group obtained a high-quality crystal structure of the (+)-α-methylbutyrate of kinamycin D. The refined data set was shown to best accommodate a diazo group, rather than a cyanamide or isonitrile function. Contemporaneously, Dmitrienko prepared a wholly synthetic derivative of prekinamycin and showed that spectroscopic data of the synthetic material were significantly different than that of natural prekinamycin. It was shown that 13C NMR and IR spectroscopic data for the kinamycins agree best with those of known diazofluorenes, such as 9-diazofluorene. The Dmitrienko group also noted that the nitrogen coupling constants of [15N2]-kinamycin D21 (JN,N = 3.4 Hz, 1JC,N = 21.2 Hz, 2JC,N = 5.4 Hz) were similar to those of [15N2]-ethyldiazoacetate (JN,N = 5.7 Hz, 1JC,N = 20.4 Hz, 2JC,N = 5.4 Hz) and that the nitrogen chemical shifts of [15N2]-kinamycin D21 (δ 241.6, 344.6) were similar to known diazo compounds (δ 227–287, δ 332–441).
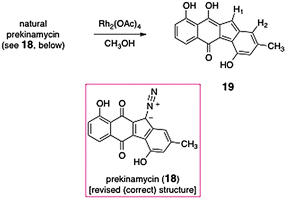
As a result of these careful investigations the structure of the kinamycins and prekinamycin were reassigned as diazotetrahydrobenzo[b]fluorenes24,25 (diazofluorenes; correct structures shown in Table 1 and Scheme 2), a substructure that, like the N-cyanoindole originally proposed, was without close analogy in natural products chemistry.
2.2 The lomaiviticins
The lomaiviticins were isolated as amorphous red powders from the fermentation broth of a bacteria originally classified as Micromonospora lomaivitiensis12 (subsequently reclassified as Salinispora).1 Analysis of the mass spectrometry data, as well as proton and carbon-13 NMR spectra, led He and co-workers to propose a C2-symmetric dimeric structure with the molecular formula of C68H80N6O24 for lomaiviticin A (11). Extensive multidimensional NMR analyses were used to establish connectivity and relative stereochemistry. The connectivity of the bis(cyclohexenone) core of lomaiviticin A (11) was determined by HMBC and HMQC analysis. A w-coupling between H-2 and H-4 in the COSY spectrum necessitated that these protons be syn-disposed (Fig. 1). The location of the bridging carbon–carbon bond was determined by HMBC and HMQC analysis: a cross-peak from H-2 to C-2/C-2′ was observed in both the HMBC and HMQC spectra, establishing the location of this key bond. The presence of carbohydrate residues – N,N-dimethylpyrrolosamine26 and oleandrose – was revealed, and the relative (but not absolute) stereochemistry of these carbohydrates, including the stereochemistry of the glycoside linkages, was elucidated by analysis of 3JH,H coupling constants. The location of the aminosugar was established by observation of an HMBC correlation from H-4 to the anomeric carbon of the aminosugar. The remaining carbohydrate was assigned to the C-3 oxygen atom, and this was supported by observation of an ROE enhancement between the C-3 ethyl substituent and the anomeric proton of the oleandrose residue. The NMR resonances of the 5,8-dihydroxy-1,4-naphthoquinone substructure were typical. On the basis of a broadened 13C NMR signal at δ 78.8, and an IR band at 2148 cm−1, a diazofluorene function was assigned. These latter data agree well with those of the kinamycins (vide supra).Analyses of HRMS, 1H, and 13C data for lomaiviticin B (12) supported the existence of a C2-symmetric structure with a molecular formula of C54H56N6O18. In this case, the ketone resonance of lomaiviticin A (11, δ 198.4) was replaced by a quaternary carbon at δ 96.5. Additionally, the oleandrose residues of lomaiviticin A (11) were absent in the spectra of lomaiviticin B (12), lending support to the cyclic furanol structure of 12. To the extent that the lomaiviticins may share similar biosynthetic pathways, the cyclic structure of lomaiviticin B (12) lends support to the relative stereochemical assignments of the bis(cyclohexenone) core of lomaiviticin A (11). The remaining diazofluorene resonances were consonant with lomaiviticin A (11) and the kinamycins. The absolute stereochemistry of the lomaiviticins was not established, but was assigned by analogy to that of the kinamycins. Both lomaiviticins are levorotatory.
3 Biosyntheses of the kinamycins
The biosyntheses of the kinamycins was studied extensively by Gould and co-workers. A comprehensive accounting of this work can be found in a recent review,15 and a brief synopsis is presented here. The pathway begins with elaboration of 10 equivalents of S-acetylcoenzyme A to the natural product dehydrorabelomycin (20, Scheme 3). A novel ring contraction then occurs to form kinobscurinone (21).27 As discussed above, labeling studies established that the diazo functional group is derived from ammonia. It was shown that this proceeds by a two-step process, via the intermediacy of the metabolite stealthin C (22).28 The mechanism of the oxidation of 22 to 18 is not known. Interestingly, owing to the presence of paramagnetic valence tautomers, both kinobscurinone (21)27 and stealthin C (22)28 are NMR silent. The intermediacy of 21 and 22 in kinamycin biosyntheses have been unequivocally established by isotope-enrichment studies.27,28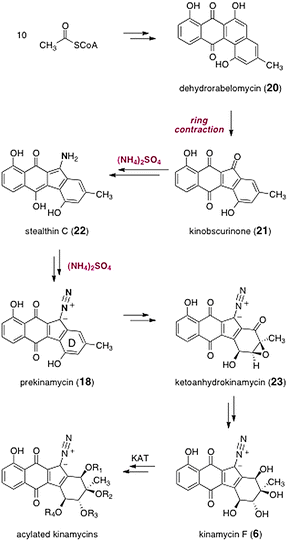 | ||
| Scheme 3 Biosyntheses of the kinamycins. | ||
Oxidative dearomatization of the D-ring then generates ketoanhydrokinamycin (23), itself a metabolite identified in fermentation broths containing the kinamycins.8 Although the intermediacy of ketoanhydrokinamycin (23) in kinamycin biosynthesis has not been rigorously established, its structural homology to, and co-production with, the kinamycins makes it a likely intermediate. Finally, a sequence of oxidation state changes and rearrangements is proposed to generate kinamycin F (6). A biomimetic approach to the kinamycins, which hinges on the transformation of synthetic constructs resembling ketoanhydrokinamycin (23), has been described by Dmitrienko and co-workers.29
A 669 kDa multifunctional enzyme (or enzyme complex), termed kinamycin acetyltransferase 1 (KAT 1), has been identified in the final steps of kinamycin biosynthesis.30 In cell-free experiments, KAT 1 exhibited dual acetyltransferase activity. It was shown to catalyze the acetyl coenzyme A-dependent conversion of the tetraol kinamycin F (6) to the monoacetate kinamycin E (5). KAT 1 also converted the latter to the diacetate kinamcyin D (4). Thus, kinamycin F (6) is a precursor to kinamycins D (4) and E (5), and potentially many other kinamycins, although the remaining KATs have yet to be identified.
4 Syntheses of the kinamycins
4.1 Enantioselective synthesis of (−)-kinamycin C (Porco and Lei)
In 2006, Porco and Lei reported a synthesis of (−)-kinamycin C (3), which constitutes the first completed route to any of the kinamycins.31 Retrosynthetically, (−)-kinamycin C (3) was deconstructed to the tetracyclic ketone 24 by manipulation of the oxidation state of the naphthoquinone and cleavage of the diazo function (Scheme 4). The tetracyclic skeleton of the ketone 24 was deconstructed by Friedel–Crafts acylation and Stille coupling reactions to form the aryl stannane 25 and the vinyl bromide 26 as synthetic precursors. The aryl stannane 25 was envisioned to arise from 2-bromojuglone (27). The vinyl bromide was derived from the acetal 28, itself ultimately prepared from an aromatic precursor.32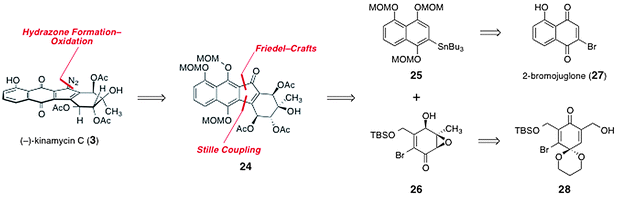 | ||
| Scheme 4 Porco's retrosynthetic analysis of (−)-kinamycin C (3). | ||
Porco's route to (−)-kinamycin C (3) began with 2,5-dihydroxybenzaldehyde (29), which was elaborated to the enone 26 by the sequence shown in Scheme 5. Bromination,33 methylation, and reduction afforded the diol 30 (79% overall). Oxidation with bis(acetoxy)iodobenzene formed the quinone monoketal 32. Protecting group manipulations then provided the acetal 33 (72%, three steps).
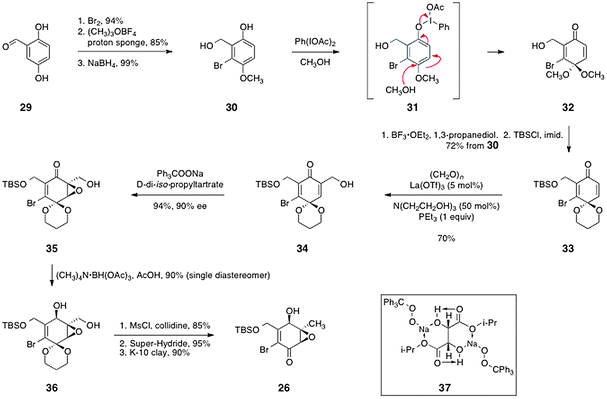 | ||
| Scheme 5 Porco's synthesis of the enone 26. | ||
The authors then sought to introduce a hydroxymethylene substituent at the unsubstituted α-position of the enone 33. This was accomplished by implementation of a modified Baylis–Hillman reaction34 using formaldehyde as the electrophile, which formed the desired product 34 in 70% yield. Asymmetric nucleophilic epoxidation35–37 then generated the epoxy alcohol 35 in 94% yield and 90% ee. In this reaction, it has been proposed that the sodium salt of tritylperoxide forms a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 complex with D-di-iso-propyl tartrate (see structure 37). It is believed that this complex is the active nucleophilic epoxidation reagent. Hydroxyl-directed reduction of the ketone function of 35 then afforded the alcohol 36 as a single diastereomer (90%). Finally, an efficient three-step sequence comprising mesylation of the primary alcohol function, reduction using lithium triethylborohydride (Super-Hydride®), and cleavage of the acetal afforded the enone 26 in 73% overall yield.
:1 complex with D-di-iso-propyl tartrate (see structure 37). It is believed that this complex is the active nucleophilic epoxidation reagent. Hydroxyl-directed reduction of the ketone function of 35 then afforded the alcohol 36 as a single diastereomer (90%). Finally, an efficient three-step sequence comprising mesylation of the primary alcohol function, reduction using lithium triethylborohydride (Super-Hydride®), and cleavage of the acetal afforded the enone 26 in 73% overall yield.
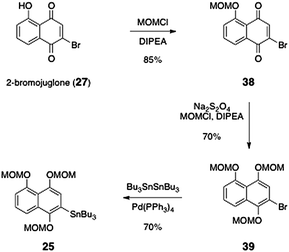
The synthesis of the arylstannane coupling partner 25 was accomplished by an efficient three-step sequence (Scheme 6). First, 2-bromojuglone (27)38 was protected as its methoxymethyl ether derivative 38 (85%). The quinone was reduced using sodium thiosulfate, and the resulting hydroquinone was protected in situ with methoxymethyl chloride, to afford the arene 39 (70%). Finally, stannylation using tetrakis(triphenylphosphine)palladium and hexabutylditin39 afforded the arylstannane coupling partner 25 in 70% yield.
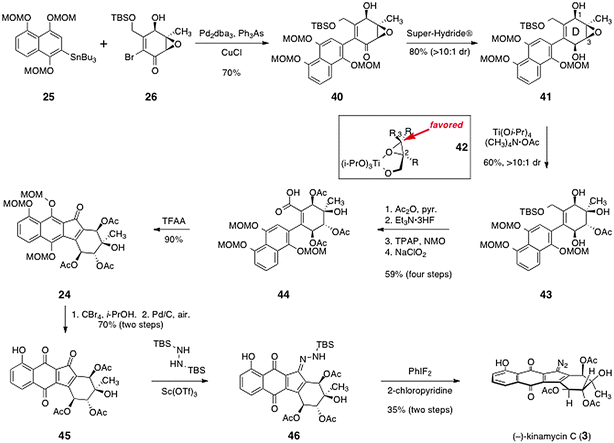
Porco's pathway to complete the synthesis of (−)-kinamycin C (3) is shown in Scheme 7. The arylstannane 25 and the α-bromoenone 26 were efficiently coupled by a Stille reaction using tris(dibenzylideneacetone) dipalladium and triphenylarsine (70%). The coupling product (40) was reduced with high diastereoselectivity using Super-Hydride®, to afford the diol intermediate 41 (80%). To establish the tertiary alcohol function of the kinamycin D-ring, the epoxide of 41 was opened regioselectively using tetramethylammonium acetate and titanium tetraisopropoxide as mediator, to afford the acetate 43 in good yield and high diastereoselectivity. These epoxide-opening conditions were originally developed by Sharpless and co-workers for the regiocontrolled opening of 2,3-epoxy alcohols.40 It was proposed that ligand exchange of the substrate with isopropoxide forms a covalently-bound substrate–titanium complex (42). Nucleophilic attack on this complex at the 3-position is favored over attack at the 2-position. In the case of 41, this model would suggest binding of the C-1 hydroxyl group to the titanium center, followed by attack at the 3-position. Under these conditions, where the weak nucleophile acetate is present, steric considerations may also favor attack at the 3-position of the epoxy alcohol functionality of 41. An added virtue of this approach is that it delivers the oxygenation with the acetate functionality required for synthesis of (−)-kinamycin C (3) in place.
 | ||
| Scheme 7 Completion of the synthesis of (−)-kinamycin C (3) by Porco and Lei. | ||
With the D-ring of the target established, Porco and Lei focused on completion of the tetracyclic framework and installation of the diazo function. Following protection of the secondary alcohols as their corresponding acetates, the silyl ether was cleaved and the resulting primary alcohol was oxidized to the carboxylic acid 44 by a two-step sequence (59%, four steps). To close the fourth and final ring, the acid 44 was treated with trifluoroacetic anhydride in dichloroethane. Under these conditions, the acid may be activated to form a mixed anhydride, which undergoes ionization to form an acylium ion that is trapped by the electron-rich aromatic ring. Rearomatization then forms the tetracyclic ketone 24 in high yield (90%).
The methoxymethyl ether protecting groups of 24 were then cleaved using carbon tetrabromide in iso-propanol. The resulting hydroquinone function was oxidized by palladium on carbon under an atmosphere of air to afford the quinone 45 (70%, two steps). Installation of the diazo function was completed by a two-step procedure. First, the ketone function of 45 was condensed with N,N′-bis(tert-butyldimethylsilyl)hydrazine in the presence of scandium triflate,41 which formed the N-tert-butyldimethylsilyl hydrazone 46. The hydrazone 46 was then oxidized using difluoroiodobenzene42 to afford (−)-kinamycin C (3) in 35% yield from 45.
4.2 Synthesis of (±)-O-methyl-kinamycin C (Ishikawa and co-workers)
In 2007, Ishikawa and co-workers reported a synthesis of (±)-O-methylkinamycin C (47).43,44 As shown in Scheme 8, the authors envisioned that 47 could be derived from the α-hydroxyindanone 48 by installation of the diazo substituent and oxidation state manipulations. The α-hydroxyindanone 48 was anticipated to arise from the enoxysilane 49. The latter could be formed by an endo-selective Diels–Alder reaction between the indenone 51 and the diene 52 (see transition state 50).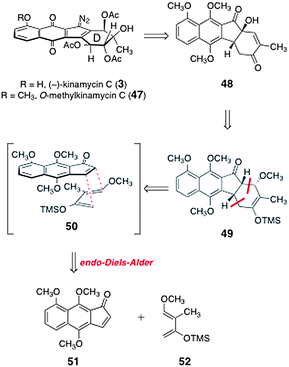 | ||
| Scheme 8 Ishikawa's retrosynthesis of (±)-O-methyl-kinamycin C (47). | ||
The synthesis of the indenone 51 is shown in Scheme 9.43 Beginning with 1,5-dihydroxynapthalene (53), acylation with acetic anhydride formed 1,5-diacetoxynapthalene (54, 89%). Oxidation using N-bromosuccinimide in a mixture of acetic acid and water38,45 formed O-acetyl-2-bromojuglone (55, 84%). The methyl ether 56 was then obtained by a two-step sequence comprising hydrolysis of the acetate ester and methylation of the resulting phenol with iodomethane in the presence of silver oxide (65%, two steps). The quinone function of 56 was reduced with stannous chloride and the resulting hydroquinone was methylated to form the aryl bromide 57 (77%, two steps). Lithium–halogen exchange followed by trapping with N,N-dimethylformamide formed the aldehyde 58 (84%). The aldehyde product 58 was then condensed with malonic acid to afford, after decarboxylation, the α,β-unsaturated carboxylic acid 59 (93%). Finally, the olefin of 59 was reduced (H2, Pd/C, 93%) and the resulting product 60 was cyclized using phosphorous pentoxide in the presence of methanesulfonic acid. The dihydroindenone so formed (not shown) was oxidized by heating with IBX in DMSO to give 51 (61%, two steps).46
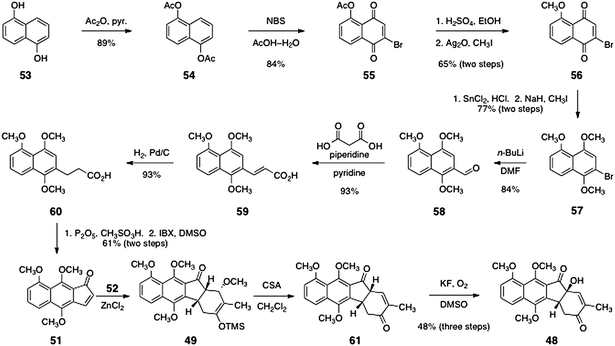 | ||
| Scheme 9 Ishikawa's synthesis of the unsaturated ketone 48. | ||
The key Diels–Alder cycloaddition was effected by treating a mixture of the indenone 51 and the diene 52 (see Scheme 8) with zinc chloride in dichloromethane at −15 °C. Under these conditions, the expected endo-adduct 49 was formed. Treatment of the adduct 49 with camphorsulfonic acid in methanol afforded the unsaturated ketone 61. Finally, the unsaturated ketone 61 was regioselectively oxygenated by treatment with potassium fluoride (0.1 equiv.) in methylsulfoxide under an atmosphere of air. This sequence generated the tertiary alcohol 48 in 48% yield over three steps.
The final steps of Ishikawa's pathway to 47 are shown in Scheme 10. First, the allylic alcohol function of 48 was oxidized by a substrate-directed dihydroxylation reaction, as described by Donohoue and co-workers,47 to afford the dihydroxylation product 62 (66%). The dihydroxylation product 62 was then persilylated to form the enoxysilane 63 (78%). Rubottom oxidation of 63 then generated the α-hydroxyketone 64. The oxidation was found to occur with selectivity for the undesired α-oxidation product. Fortunately, however, this product epimerized to the desired β-epimer 65 on standing (67–71% from 63). The silyl ether functions of 65 were then cleaved by treatment with aqueous methanol to form the tetraol 66 (91%). The secondary hydroxyl groups of 66 were selectively acylated to form the diacetate 67 (70%). The cyclohexanone function of 67 was then reduced by a diastereoselective reduction using tetramethylammonium triacetoxyborohydride.48 Under these conditions, competitive reduction of the cyclopentanone carbonyl was also observed. Fortunately, this carbonyl could be restored by treatment with manganese dioxide, affording the desired reduction product 69 in 69% yield as a 5:1 mixture of diastereomers. The major diastereomer 69 likely arises from intramolecular hydride delivery of an in situ generated borate (see structure 68).
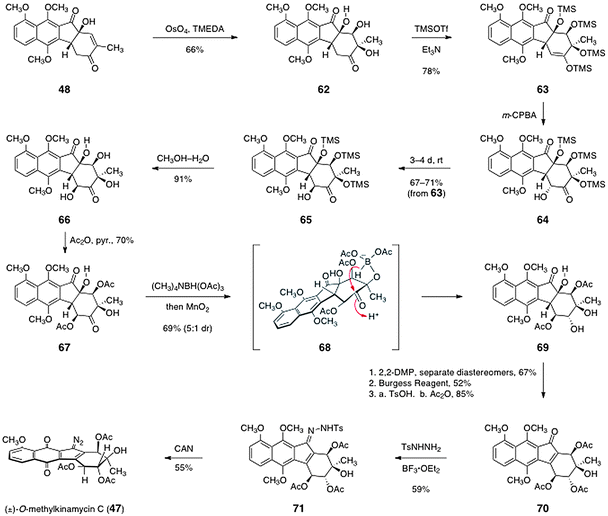 | ||
| Scheme 10 Completion of the synthesis of (±)-O-methylkinamycin C (47) by Ishikawa and co-workers. | ||
The synthesis of 47 was completed by the following sequence. First, the vicinal diol function of 69 was protected as the corresponding acetonide derivative (67%, not shown). This protection step allowed for the separation of the minor diastereomer formed in the preceding reduction. Following separation, the tertiary hydroxyl group was dehydrated to form an unsaturated ketone (52%, not shown). The acetonide was then cleaved and the secondary hydroxyl groups were acylated to generate the triacetate 70 (85%). Finally, the tosylhydrazone 71 was formed by treatment of 70 with para-toluenesulfonylhydrazine and boron trifluoride etherate (59%). The hydrazone 71 was then oxidized using ceric ammonium nitrate. Under these conditions, the methyl ethers of the hydroquinone function were also cleaved49 to afford 47 directly (55%).
4.3 Enantioselective syntheses of kinamycins C, F, and J (Nicolaou and co-workers)
In 2007, Nicolaou and co-workers reported efficient enantioselective syntheses of (−)-kinamycin C (3), (−)-kinamycin F (6), and (−)-kinamycin J (10).50 The authors envisioned that the α-hydroxyketone 72 could serve as a precursor to the targets (Scheme 11). The α-hydroxyketone function of 72 was envisioned to arise from an Ullmann coupling–benzoin condensation sequence between the ortho-bromoaryl aldehyde 73 and the α-iodoenone 74. These were derived from the bromojuglone derivative 75 and the enantiomerically enriched enone 76, respectively.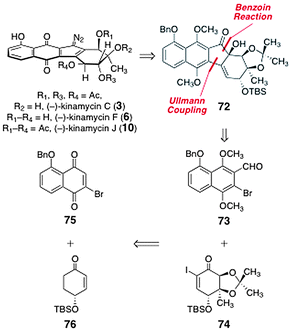 | ||
| Scheme 11 Retrosyntheses of (−)-kinamycin C (3), (−)-kinamycin F (6), and (−)-kinamycin J (10) by Nicolaou and co-workers. | ||
The ortho-bromoaryl aldehyde 73 was prepared by an efficient five-step sequence (Scheme 12). Beginning with 2-bromojuglone (27), radical allylation afforded the allylquinone 77 (75%). O-Benzylation formed the benzyl ether 78 (92%). Reduction and in situ methylation provided the hydroquinone dimethyl ether 79 (82%). Isomerization of the alkene was effected by treatment of 79 with potassium tert-butoxide (98%). The resulting styrene derivative (not shown) was subjected to oxidative cleavage to afford the ortho-bromoaryl aldehyde 73 (84%).
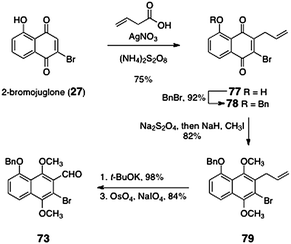 | ||
| Scheme 12 Synthesis of the ortho-bromoaryl aldehyde 73 by Nicolaou and co-workers. | ||
Nicolaou's synthesis of the α-iodoenone 74 is shown in Scheme 13. The synthesis began with (R)-4-(tert-butyldimethylsilyloxy)cyclohex-2-ene-1-one (76), prepared in >80% ee by desymmetrization of the corresponding ketone.51 Copper-catalyzed 1,4-addition of methyl magnesium bromide, trapping with chlorotrimethylsilane, and reoxidation then generated the β-methylcyclohexenone 80 (90%). This product (80) was subjected to a diastereoselective catalytic dihydroxylation to afford the diol 81 (76%). The diol 81 was readily recrystallized to >98% ee. Protection of the diol function afforded the acetonide 82 (95%). Generation of the enoxysilane of 82 followed by palladium-mediated oxidation then afforded the enone 83 (84%). Finally, the enone 83 was iodinated under Johnson conditions52 to provide the target α-iodoenone 74 (92%).
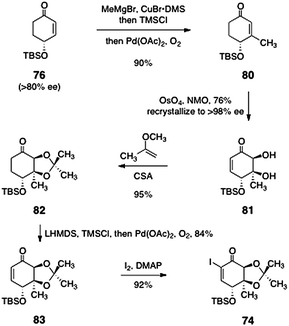 | ||
| Scheme 13 Nicolaou's synthesis of the α-iodoenone 74. | ||
The coupling of the aryl aldehyde 73 and the α-iodoenone 74 was affected by a modified Ullmann reaction (83%, Scheme 14).53 The resulting arylation product (84) was subjected to a benzoin condensation using the triazole catalyst 85,54,55 to form the benzoin product 86 (78%). Acylation of the tertiary alcohol function of 86 then afforded the acetate 87 (95%).
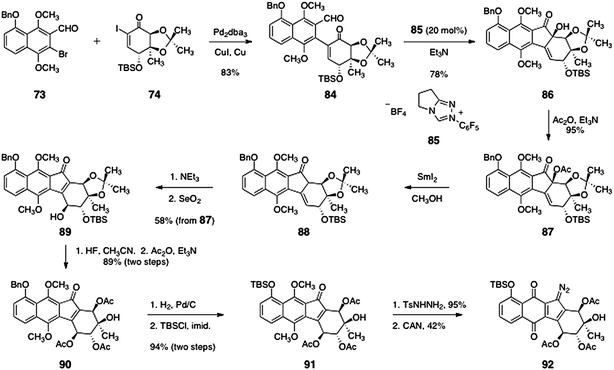 | ||
| Scheme 14 Nicolaou's synthesis of the penultimate precursor 92. | ||
The α-acetoxyketone function of 87 was reduced with samarium diiodide,56,57 to afford the β,γ-unsaturated ketone 88. Treatment with triethylamine resulted in smooth isomerization of the olefin to form an α,β-unsaturated ketone (not shown). Diastereoselective γ-oxidation (selenium dioxide) generated the secondary alcohol 89 (58%, three steps). The acetonide and silyl ether functions of 89 were cleaved by treatment with hydrofluoric acid to afford a tetraol intermediate (not shown) that was regioselectively acylated at the secondary alcohol functions, providing the triacetate 90 in 89% yield (two steps). Hydrogenolysis of the benzyl ether of 90 followed by silylation of the resulting phenol afforded the key silyl ether 91 (94%). Finally, condensation of 91 with para-toluenesulfonylhydrazine afforded a hydrazone (95%, not shown) that was oxidized with ceric ammonium nitrate to form the diazofluorene 92 (42%).
The key diazofluorene 92 was efficiently transformed to (−)-kinamycin C (3), (−)-kinamycin F (6), and (−)-kinamycin J (10, Scheme 15). Cleavage of the silyl ether of 92 (aqueous hydrochloric acid) afforded (−)-kinamycin C (3, 95%). Alternatively, treatment of 92 with lithium hydroxide resulted in liberation of the phenol function and saponification of the acetate esters, providing (−)-kinamycin F (6) in 92% yield. Finally, acylation of the tertiary hydroxyl of 92 (acetic anhydride, triethylamine), followed by cleavage of the silyl ether (hydrochloric acid), provided (−)-kinamycin J (10, 80%, two steps).
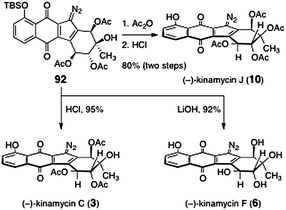 | ||
| Scheme 15 Completion of the syntheses of (−)-kinamycin C (3), (−)-kinamycin F (6), and (−)-kinamycin J (10) by Nicolaou and co-workers. | ||
4.4 Enantioselective synthesis of (−)-kinamycin F (Herzon and co-workers)
In 2010, Herzon and co-workers reported a synthesis of (−)-kinamycin F (6).58 It was envisioned that (−)-kinamycin F (6) could be prepared from the protected diazofluorene 93 (Scheme 16). The diazofluorene 93 was formed by diazo transfer to the triacylcyclopentadiene 94 (it was later observed that 94 existed as the hydroxyfulvene tautomer, vide infra). The triacylcyclopentadiene 94 was derived from cyclization of the γ-quinonyl enone 95. Intermediate 95 was synthesized by coupling of O-(methoxymethyl)-2-bromo-3-methoxyjuglone (96) and the β-trimethylsilylmethyl-α,β-unsaturated ketone 97. The latter two intermediates were prepared from juglone (98) and the silyl ether 99, respectively.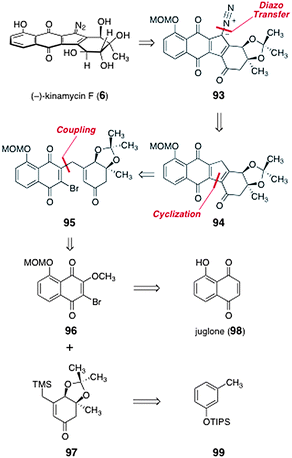 | ||
| Scheme 16 Retrosynthesis of (−)-kinamycin F (6) by Herzon and co-workers. | ||
The synthesis of the O-(methoxymethyl)-2-bromo-3-methoxyjuglone (96) is shown in Scheme 17. Bromination of juglone (98) afforded the dibromojuglone derivative 100. The phenol function of 100 was protected as its methoxymethyl ether 101 (50%, two steps). Heating mixtures of 101 and sodium carbonate in methanol resulted in regioselective substitution of the C-3 bromide substituent to form O-(methoxymethyl)-2-bromo-3-methoxyjuglone (96, 96%).
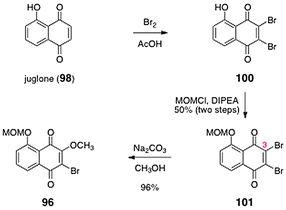 | ||
| Scheme 17 Synthesis of O-(methoxymethyl)-2-bromo-3- methoxyjuglone (96). | ||
The synthesis of the β-trimethylsilylmethyl-α,β-unsaturated ketone 97 began with the silyl ether 99 (prepared in quantitative yield by silylation of meta-cresol, Scheme 18). Birch reduction of 99 formed the cyclohexadiene derivative 102 (>99%). Regio- and stereoselective dihydroxylation59 of 102 afforded the diol 103 (55%). The enantiomeric excess of the product was modest (66%), but a subsequent intermediate (105) could be recrystallized to >95% ee. The diol 103 was transformed to the acetonide 104 by treatment with 2,2-dimethoxypropane and pyridinium para-toluenesulfonate (88%). Next, the enone function was installed by sequential α-selenylation, peroxide oxidation, and elimination (57%, two steps).60,61 Finally, the unsaturated ketone 105 was homologated by copper-catalyzed 1,4-addition of trimethylsilylmethyl magnesium chloride, trapping with chlorotrimethylsilane, and reoxidation, to afford 97 (88%).
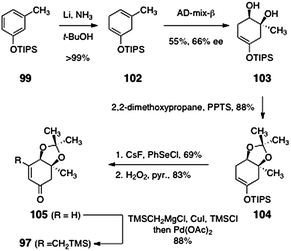 | ||
| Scheme 18 Synthesis of the β-trimethylsilylmethyl-α, β-unsaturated ketone 97. | ||
Treatment of a mixture of 96 and 97 with tris(diethylaminosulfonium) trimethyldifluorosilicate [TASF(Et)] resulted in smooth addition–elimination to the naphthoquinone, forming the γ-alkylation product 95 (79%, Scheme 19). Heating of the γ-alkylation product 95 in the presence of palladium acetate, polymer-supported triphenylphosphine, and silver carbonate provided the cyclised product 94 (66%). The product 94 was shown to exist in the hydroxyfulvene tautomer 106. Treatment of 106 with Hünigs base and trifluoromethanesulfonyl azide62 resulted in efficient diazo transfer, providing the diazofluorene 93 in 99% yield. Under these conditions, the hydroxyfulvene is deprotonated to generate a cyclopentadienyl anion. Attack of this anion on the triflyl azide reagent provides the observed product (93). It is noteworthy that although the anionic charge of the conjugate base of 106 is delocalized over the entire cyclopentadiene ring, only diazotransfer to the desired position generates a stable product. One of the earliest applications of para-toluenesulfonyl azide, by von Doering, was in the synthesis of diazocyclopentadiene itself.63 In the present setting, only the highly electrophilic diazotransfer reagent triflyl azide was found to react with the conjugate base of 106.
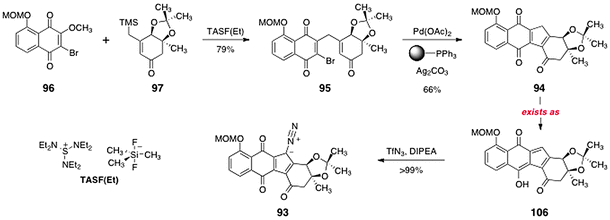 | ||
| Scheme 19 Synthesis of the diazofluorene 93. | ||
The synthesis of (−)-kinamycin F (6) was completed by the pathway shown in Scheme 20. The acetonide protecting group of 93 hinders the approach of reagents from the β-face, and this bias was used to establish the trans-1,2-diol function of (−)-kinamycin F (6). First, the enoxysilane 107 was formed by treatment with tri-iso-propylsilyl trifluoromethanesulfonate and Hünig's base. Addition of dimethyldioxirane resulted in oxygen atom transfer to the α-face of 107 (see 108), to form a putative silyloxyepoxide intermediate 109. By employing methanol as co-solvent, 109 was cleaved in situ to form the free α-hydroxy ketone 110 (76%). Treatment of 110 with borane–tetrahydrofuran complex at −78 °C, followed by warming to −20 °C, resulted in smooth reduction of the ketone to form the trans-1,2-diol 112 (58%). It was proposed that this reduction proceeds via the in situ-generated borinic ester 111. Global deprotection (methanol, hydrogen chloride) then afforded (−)-kinamycin F (6, 65%).
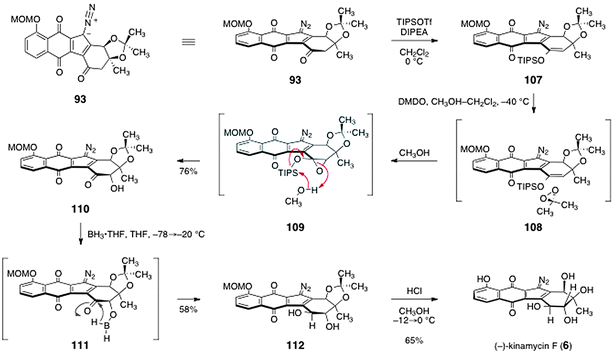 | ||
| Scheme 20 Completion of the synthesis of (−)-kinamycin F (6) by Herzon and co-workers. | ||
5 Synthetic studies toward the lomaiviticins
5.1 Introduction
Significant progress toward the syntheses of the lomaiviticins has been achieved, although their complete total synthesis remains an ongoing challenge. Here we review efforts toward the synthesis of the dimeric core of the lomaiviticins, syntheses of advanced dimeric intermediates, synthesis of the monomeric lomaiviticin aglycon, synthesis of the lomaiviticin aglycon, and finally methods for construction and introduction of the 2,6-dideoxyglycoside residues of the lomaiviticins.The lomaiviticins contain the densely-functionalized bis(cyclohexene) cores 113 and 114, respectively (Fig. 3). The syntheses of these substructures present considerable challenges, and was the focus of most early work. At least two significant obstacles are identified. The first involves the stereocontrolled construction of the carbon–carbon bonds (blue in 113 and 114) that bridge the two halves of each core structure. These bonds are exceedingly hindered (C-3/C-3′ are tetrasubstituted) and the inherent facial selectivity in reactions that might be useful for their construction is ambiguous. The second challenge derives from the substitution about the cyclohexene rings. In the lomaiviticin A core 113, these rings contain a β-hydroxyketone substructure, which is susceptible to an elimination pathway (see 115 → 116, Scheme 21). The elimination product 116 can tautomerize to a hydroquinone ether 117, effectively eliminating all stereocenters in this ring. Somewhat ironically, the steric hindrance that makes formation of the bridging carbon–carbon bond difficult may afford some level of protection against this pathway in late-stage dimeric intermediates. Conversely, monomeric intermediates (i.e., those that lack the bridging C–C bond), which possess much more accessible α-hydrogen atoms, are expected to exhibit heightened reactivity toward this mode of decomposition.
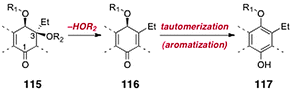 | ||
| Scheme 21 The β-elimination–aromatization pathway. | ||
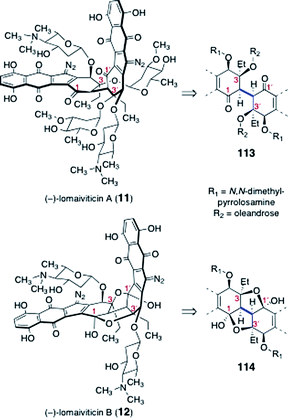 | ||
| Fig. 3 Lomaiviticins A (11) and B (12), and their respective dimeric cores (113, 114). | ||
5.2 Syntheses of dimeric core structures (Nicolaou and co-workers)
In order to address the challenges posed by the dimeric core of the lomaiviticins, Nicolaou and co-workers targeted the synthetic constructs 118 and 119 (Scheme 22).64 It was envisioned that 118 and 119 could be prepared from the tricycle 120. In their synthetic plan, X is a substituent that may serve to bridge the two rings and, in subsequent transformations, undergo conversion to a dicarbonyl function. The construct 120 was simplified to the diketone 121. The latter was anticipated to arise from the bis(cyclohexenone) 122, itself formed by reductive coupling of 2-iodo-cyclohex-2-ene-1-one (not shown).65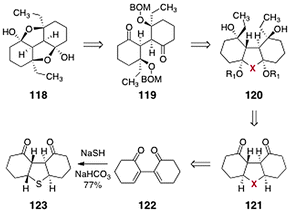 | ||
| Scheme 22 Retrosynthesis of 118 and 119 by Nicolaou and co-workers. | ||
In preliminary studies, the suitability of a sulfur-based bridge (see 123) was investigated. Intermediate 123 was formed in a single-step and 77% yield by double 1,4-addition of sodium hydrogen sulfide to 122. A malonate-based strategy later proved superior; the development of this approach is shown in Scheme 23. Conjugate addition of dimethyl malonate to 122 yielded the double-addition product 124 as a single diastereomer (85%). Treatment of 124 with excess ethyl magnesium bromide in the presence of cerium chloride66 triggered a cascade reaction, resulting in formation of the product 126 directly (57%). The authors postulated that 126 forms by a sequence comprising addition of the organometal reagent to the ketone functions of 124, cyclization to form a tetracyclic lactone (125), and cleavage of this lactone by an organometal addition–retro-aldol cascade.
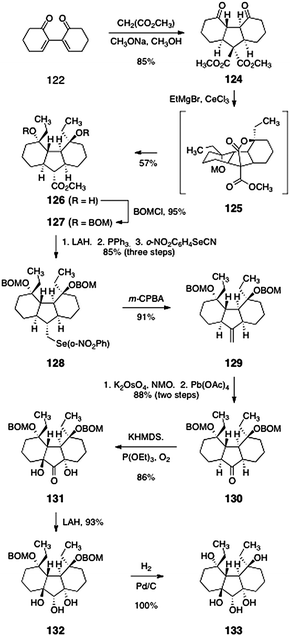 | ||
| Scheme 23 Synthesis of the tricycle 133 by Nicolaou and co-workers. | ||
Protection of the alcohol functions of 126 as their benzyloxymethyl ethers proceeded smoothly to provide 127 (95%). Reduction of the methyl ester (lithium aluminium hydride) formed a primary alcohol (not shown). Iodination (triphenylphosphine, iodine) and displacement provided the ortho-nitrobenzeneselenide 128 (85%, three steps). Selenoxide formation (m-CPBA) and in situ elimination furnished the alkene 129 (91%). A two-step oxidative cleavage [K2OsO4, NMO, then Pb(OAc)4] provided the ketone 130 (88%, two steps). Double α-hydroxylation of the ketone 130 (potassium hexamethyldisilazide, triethylphosphite, dioxygen) furnished the C2-symmetric diol 131, as a single diastereomer (86%). Stereoselective reduction of the ketone function (lithium aluminium hydride) yielded the triol 132 (93%). Finally, hydrogenolysis of the benzyloxymethyl ether functions afforded the pentaol 133 (100%).
The completion of the lomaiviticin A core structure 119 is shown in Scheme 24. First, the triol 132 was cleaved (lead tetraacetate) to afford the cyclic hemiacetal 134 (90%). In this transformation, it was postulated that initial oxidation occurs to form a ketoaldehyde (not shown). This intermediate then undergoes spontaneous cyclization to form the observed product (134). In the next step, reduction of the latent carbonyl functions of 134 (lithium borohydride) and oxidative cleavage provided the hemiketal 135 (66%, two steps, inconsequential 3:1 mixture of diastereomers). The product 135 was then oxidized (TPAP, NMO) to afford the lomaiviticin A core structure 119 (55%).
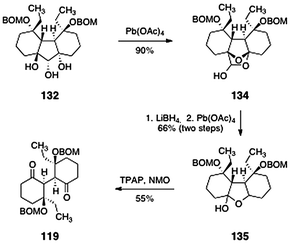 | ||
| Scheme 24 Completion of the synthesis of the lomaiviticin A core 119 by Nicolaou and co-workers. | ||
The synthesis of the lomaiviticin B core 118 is shown in Scheme 25. Oxidation of 133 (lead tetraacetate) produced the monoformate 138 (95%). This transformation was postulated to proceed via the 136 and 137. Deformylation (ammonia, methanol) then afforded the fused the lomaiviticin B tetracyclic core model system 118 (40%), presumably through the intermediacy of the diketone 139.
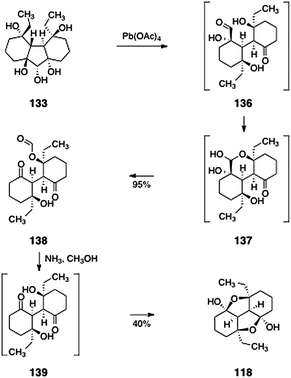 | ||
| Scheme 25 Completion of the synthesis of the lomaiviticin B core 118 by Nicolaou and co-workers. | ||
5.3 Synthesis of the C-3/C-3′-dideoxy lomaiviticin core (Sulikowski and co-workers)
Sulikowski and co-workers described an efficient approach to the bis(cyclohexenone) core of lomaiviticin A.67 The authors targeted the dimeric construct 140 (Scheme 26), which was envisioned to serve as a precursor to the lomaiviticins. The target (140) was anticipated to arise from the bis(enone) 141. The latter in turn could be synthesized by substrate-controlled stereoselective functionalization of 142. Intermediate 142 was anticipated to derive from (−)-quinic acid (143).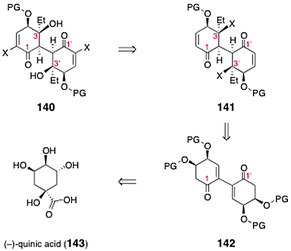 | ||
| Scheme 26 Retrosynthesis of the lomaiviticin dimeric core 140 by Sulikowski and co-workers. | ||
Sulikowski's studies began with the cyclohexenone 144 (Scheme 27) prepared from (−)-quinic acid (143) by an established sequence.68 Iodination52 then provided the α-iodoenone 145 (63%). Nickel-catalyzed reductive dimerization65 of the α-iodoenone formed the dimeric product 146 (64%). Double 1,4-addition of vinyl cuprate occurred with complete stereoselectivity, anti to the acetonide protecting groups, to form the 1,4-addition product 147 (67–75%). Protonation of the putative enolate intermediates also occurred stereoselectively, establishing the C-2/C-2′ centers of the target. The acetonide functions of 147 could be efficiently cleaved by treatment with DBU. However, under these conditions epimerization of one of the C-2 centers was observed, resulting in isolation of the C1-symmetric diol 148 (56%). Remarkably, DBU-mediated acetonide cleavage of the reduced product 149 (formed in 85% yield by catalytic hydrogenation of 147) produced an entirely different outcome. In this case, the bridged ketal 150 was isolated (49%). It is possible that 150 forms by a sequence comprising base-mediated acetonide cleavage, 1,2-addition of the resulting alcohol to the adjacent ketone, and 1,4-addition of the intermediate hemiketal.
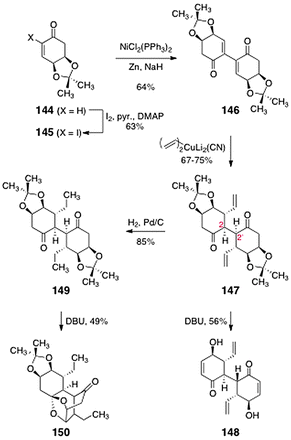 | ||
| Scheme 27 Studies toward the lomaiviticin core by Sulikowski and co-workers. | ||
In light of these results, the authors examined alternate methods to introduce the ethyl chain and cleave the acetonide function. Toward this end, the allylated bis(enoxysilane) 151 was synthesized by addition of allyltributyltin to 146 in the presence of tert-butyldimethylsilyl trifluoromethanesulfonate (97%, Scheme 28). A two-step oxidative cleavage of the alkene functions, followed by reduction (sodium borohydride) formed the diol 152 (75%, three steps). The alcohol functions of 152 were removed by mesylation, displacement with iodide, and catalytic hydrogenation, to afford 153 (55%, three steps). Treatment of 153 with tetrabutylammonium fluoride resulted in cleavage of the enoxysilane and acetonide functions. The resulting enone (not shown) was acylated and then α-iodinated to form the bis(α-iodoenone) 154 (55%, three steps). This construct constitutes the C-3/C-3′-dideoxy core of the lomaiviticins.
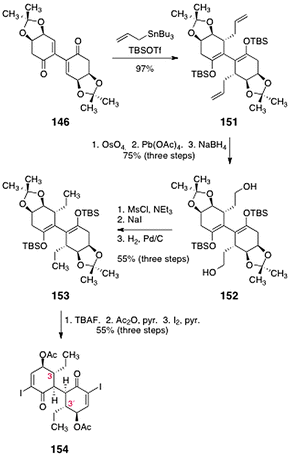 | ||
| Scheme 28 Synthesis of the C-3/C-3′-dideoxy lomaiviticin core 154 by Sulikowski and co-workers. | ||
Finally, introduction of the C-3/C-3′ hydroxyl substituents was investigated (Scheme 29). The authors envisioned that oxidation of the tetraacetate 155 (itself prepared from 151 in two steps) might form an extended silyloxycarbonium ion (157). Intramolecular trapping by the pendant acetate (158), followed by hydrolysis, would generate the C-3/C-3′-dioxygenated product 159. Unfortunately, attempted oxidation of 155 only led to the ene-1,4-dione 156 (68%).
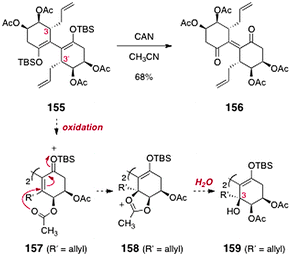 | ||
| Scheme 29 Attempted formation of the fully oxygenated lomaiviticin core by Sulikowski and co-workers. | ||
5.4 Synthesis of the complete lomaiviticin carbon skeleton (Shair and co-workers)
Over the period 2008–2010, Shair and co-workers unveiled a strategy for the synthesis of the dimeric core of the lomaiviticins69 and advanced intermediates containing the entire carbon scaffold of the natural products.70Shair's synthetic approach hinges on the oxidative coupling of monomeric intermediates. As discussed above, although a direct oxidative coupling of two synthetic “lomaiviticin monomers” (2 × 160 → 161, Scheme 30) is conceptually attractive, in practice such an approach is complicated by issues of stereocontrol and β-elimination (see also Section 5.1). To address these issues, Shair and co-workers targeted intermediates such as 163, which contain the 7-oxanorbornanone substructure. As a consequence of the fused bicyclic geometry of 163, carbon–carbon bond formation is expected to occur from the less-hindered, exo face (indicated by the blue arrow), providing the stereochemistry required for lomaiviticin synthesis. Moreover, the β-carbon–oxygen antibonding orbital is poorly aligned with the π-system of the enolate 163. It was therefore expected that β-elimination from this system would be slow.
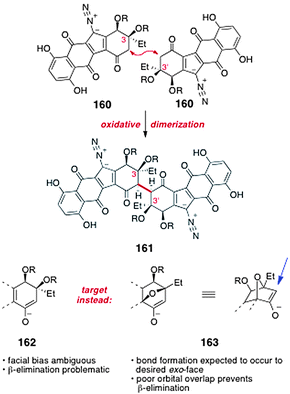 | ||
| Scheme 30 Oxidative coupling of lomaiviticin monomers 160 and formation of a 7-oxanorbornanone intermediate to circumvent β-elimination and control stereochemistry (Shair and co-workers). | ||
Initial studies targeted the core system 173 (Scheme 31).69 Michael addition of the lithium enolate of furanone 16571 to oxazolidinone 16472 provided the 1,4-addition product 166 in 90% yield (1:1 mixture of diastereomers, inconsequential). Acylation of the furanone function generated the acetoxyfuran 167 (92%). An Evans magnesium-catalyzed anti-aldol reaction73 between the acetoxyfuran 167 and β-phenylthioacrolein furnished the anti-aldol adduct 168 (60–77%). The aldol adduct 168 was protected as its silyl ether (78%, not shown), and the acetoxy group was cleaved by treatment with potassium cyanide, to afford the furanone 169 (70–95%). Oxidation of 169 (m-CPBA) generated a vinyl sulfone (not shown), which upon heating to 50 °C underwent an intramolecular Diels–Alder reaction (potentially via the furanol tautomer) to form the endo-adduct 170 as the major diastereomer (53%). Acid-mediated cleavage of the imide and silyl substituents, followed by reprotection of the secondary alcohol and saponification of the silyl ester formed the carboxylic acid 171 (71% overall). Barton decarboxylation then provided the target 173 (64%, two steps).
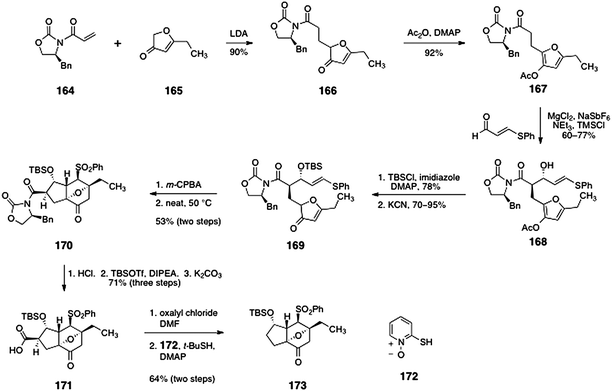 | ||
| Scheme 31 Synthesis of the tricyclic intermediate 173 by Shair and co-workers. | ||
Oxidative dimerization of 173 was realized by enolization with lithium hexamethyl-disilazide followed by addition of ferrocenium hexafluorophosphate (Scheme 32, 45–51%). The stereochemistry of the single C2-symmetric product that formed (174) was established by observation of an NOE between the α-hydrogen and the phenylsulfonyl substituent. The dimer 174 was then desilylated and the resulting alcohols were oxidized with the Dess–Martin periodinane (26%, two steps). Interestingly, the product 175 was shown to incorporate one equivalent of water, to form a stable cyclic hydrate. Finally, the oxanorbornane system of 175 was cleaved by treatment with potassium carbonate in methanol. Under these conditions, the phenylsulfonyl substituent also under went displacement by methoxide to generate the bis(methyl ether) 176 (14%). Attempts to dehydrate 176 to form the core of lomaiviticin B were unsuccessful. The authors postulated that the stability of the hydrate 176 may arise from the vinylogous diketone nature of the system, which is expected to drive the ketone–hydrate equilibrium toward the hydrate form.
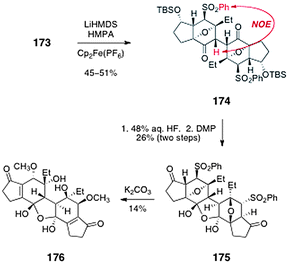 | ||
| Scheme 32 Dimerization of 173 and elaboration to 176 by Shair and co-workers. | ||
The same group extended this strategy to the first synthesis of the lomaiviticin aglycon carbon skeleton.70 As before, an oxidative dimerization of an oxanorbornanone substrate is the key step in the sequence. These studies began with the norbornanone 170, which was synthesized by the route shown in Scheme 31. Treatment of 170 with sodium borohydride in allyl alcohol as solvent resulted in diastereoselective reduction of the ketone and cleavage of the imide, to form the allyl ester 177 (89%, Scheme 33). Protection of the secondary alcohol as its pivalate ester (not shown) and cleavage of the silyl ether formed the alcohol 178 (91%, two steps). In the next step, the secondary alcohol was oxidized with dimethylsulfoxide–oxalyl chloride, which formed an α-chloro-β-ketoester (not shown). The chlorine substituent could be readily removed by treatment with zinc in acetic acid to afford the desired β-keto ester 179 in 96% yield (two steps). Treatment of the β-ketoester 179 with bis(triphenylphosphine)-palladium dichloride and tributyltin hydride resulted in decarboxylative deallylation, to provide the ketone 180 (85%). α-Selenation, oxidation, and cheletropic elimination generated the α,β-unsaturated ketone 181 (73%).
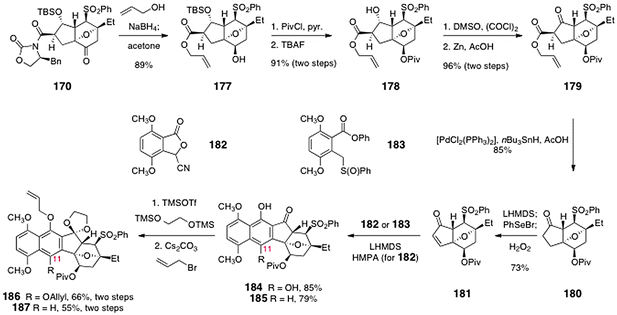 | ||
| Scheme 33 Elaboration of 170 to the annulated products 186 and 187 by Shair and co-workers. | ||
In initial studies, the unsaturated ketone was treated with the lithiated cyanophthalide 182,74 to afford the annulated product 184 (85%). Protecting group manipulations provided the acetal 186 (66%, two steps).
Sequential pivalate deprotection and oxidation (TPAP, NMO) afforded the oxidative coupling substrate 188 (90%, two steps, Scheme 34). Unfortunately, all attempts to dimerize 188 were unsuccessful. The authors reasoned that the desired product 189 may suffer destabilizing non-bonded interactions, which may inhibit approach of the monomers in the oxidative coupling reaction (Fig. 4).
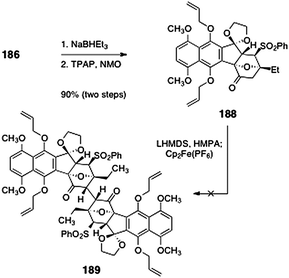 | ||
| Scheme 34 Attempted dimerization of 188 by Shair and co-workers. | ||
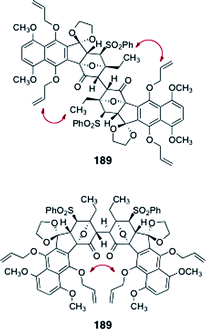 | ||
| Fig. 4 Steric interactions postulated to prevent the dimerization of 188. | ||
In order to alleviate these steric interactions, the group pursued the synthesis of 187 (Scheme 33), which lacks the C-11-allyloxy substituent. Hauser annulation75 of 181 with the sulfoxide 183 provided the C-11-deoxy product 185 (79%). Protecting group manipulations then gave 187 (55%, two steps).
Sequential pivalate reduction and oxidation provided the deoxy dimerization precursor 190 (90%, two steps, Scheme 35). Oxidative dimerization of 190 proceeded in excellent yield to afford the desired C2-symmetric dimer 191 (80%, 830 mg scale). The allyl ether functions of the dimer 191 could be efficiently cleaved by treatment with bis(triphenylphosphine)palladium dichloride and tri-butyltin hydride (90%). X-ray analysis of the product 192 confirmed that it possessed the stereochemistry required for lomaiviticin synthesis. The successful dimerization of 190 compared to 188 highlights the large degree of steric congestion in these molecules and the unexpected consequences of these interactions.
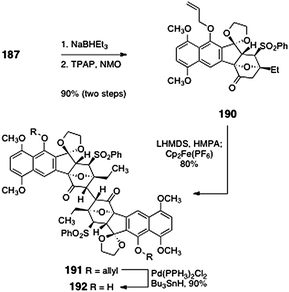 | ||
| Scheme 35 Successful dimerization of 190 to form the entire lomaiviticin carbon skeleton 191 by Shair and co-workers. | ||
5.5 Synthesis of the monomeric unit of the lomaiviticin aglycon (Nicolaou and co-workers)
Nicolaou and co-workers report the first synthesis of the monomeric unit of the lomaiviticin aglycon (193).76 Retrosynthetically, the target 193 was envisioned to arise from the ortho-bromoaryl aldehyde 194 and the α-iodoenone 195 (Scheme 36). The pathway to 193 shares some parallels to the authors' route to (−)-kinamycin C (3), (−)-kinamycin F (6), and (−)-kinamycin J (10),50 although several modifications were required to accommodate the structural elements of 193. In the course of these studies, a novel alcohol transposition reaction was discovered.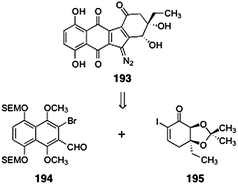 | ||
| Scheme 36 Retrosynthesis of the monomeric lomaiviticin aglycon 193 by Nicolaou and co-workers. | ||
The ortho-bromoaryl aldehyde 194 was prepared from the known intermediate 196 (Scheme 37).50 Aluminium trichloride-mediated debenzylation (80%), followed by oxidation formed a dimethoxynaphthoquinone intermediate (97%, not shown). Reduction of the quinone and protection of the resulting hydroquinone as its bis(2-trimethylsilylethoxymethyl) (SEM) ether formed the target 194 (92%).
 | ||
| Scheme 37 Synthesis of the ortho-bromoaryl aldehyde 194 by Nicolaou and co-workers. | ||
The α-iodoenone 195 was prepared by an efficient four-step sequence (Scheme 38). First, 3-ethyl-cyclohex-2-ene-1-one (197) was subjected to a Sharpless asymmetric dihydroxylation reaction, which afforded a syn-1,2-diol intermediate (not shown) in 69% yield and >95% ee. Protection of the diol function (pyridinium para-toluenesulfonate, 2-methoxypropene) formed the acetonide 198 (94%). Treatment of 198 with trimethylsilyl trifluoromethane-sulfonate and triethylamine formed an enoxysilane (not shown), that was oxidized (palladium acetate, dioxygen) to provide the enone 199 (83%). Iodination52 of the enone furnished 195 (91%).
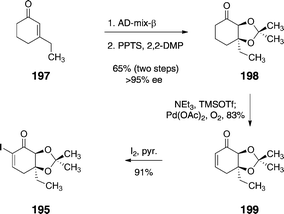 | ||
| Scheme 38 Synthesis of the α-iodoenone 195 by Nicolaou and co-workers. | ||
The ortho-bromoaryl aldehyde 194 and the α-iodoenone 195 were elaborated to the monomeric unit of the lomaiviticin aglycon (193) by the sequence outlined in Scheme 39. Palladium-mediated cross-coupling between the 194 and 195 furnished α-arylketone 200 (69%). Benzoin condensation of 200 using the catalyst 8555 furnished the α-hydroxyketone 202 (70%), as an inconsequential mixture of diastereomers (3:1). Substrates lacking the C-5 substituent gave predominantly the 1,4-addition (Stetter) product in the same transformation. Calculations suggested the bulky C-5 SEM group gears the cross-coupling product toward conformation 201, via a non-bonded interaction with the adjacent methoxy substituent. In the next step, the α-hydroxyketone function of 202 was reduced using samarium diiodide. Bubbling of dioxygen through the reaction mixture, followed by reductive work-up, afforded the γ-hydroxy-α,β-unsatured ketone 203 (76%, 1.5:1 inconsequential mixture of diastereomers). It was proposed that this novel transformation proceeds by reductive elimination of the α-hydroxyl function to form an extended samarium enolate. Regioselective addition of dioxygen at the γ-position, and subsequent reduction of the resulting alkyl hydro peroxide intermediate then affords the observed product 203. Condensation of 203 with para-toluenesulfonylhydrazine formed an intermediate hydrazone (91%, not shown). Exhaustive oxidation with the Dess–Martin periodinane (5 equiv.) produced the diazofluorene 204 (62%). Reduction of the quinone function of 204 and acylation of the resulting hydroquinone proceeded smoothly (91%). Deprotection of the acetonide (trimethylsilyl trifluoromethanesulfonate) generated the diol 205 (96%). Finally, oxidative cleavage of the hydroquinone dimethyl ether function (CAN) and saponification of the acetate protecting groups (KOH) furnished the monomeric lomaiviticin aglycon (193, 91%, two steps).
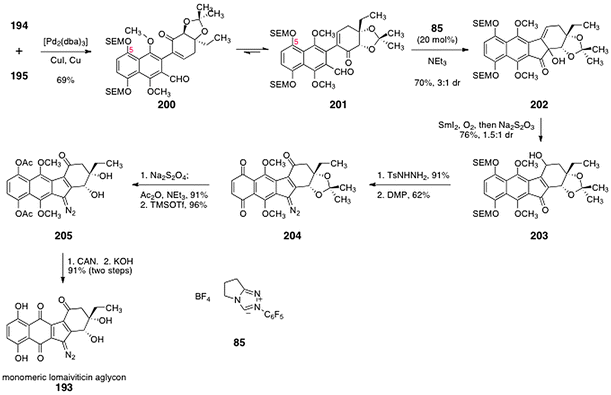 | ||
| Scheme 39 Synthesis of the monomeric lomaiviticin aglycon 193 by Nicolaou and co-workers. | ||
5.6 Synthesis of the lomaiviticin aglycon (Herzon and co-workers)
Herzon and co-workers reported a synthesis of the lomaiviticin aglycon (206).77 The lomaiviticin aglycon (206) was prepared from the protected C2-symmetric dimer 207 (Scheme 40). The latter was synthesized by dimerization of the monomeric diazofluorene 208.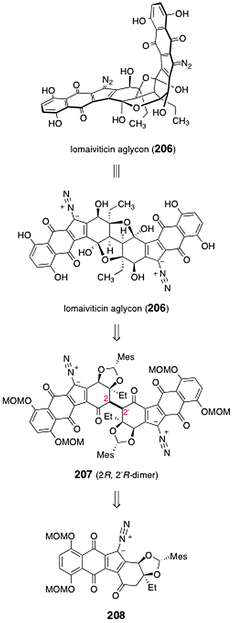 | ||
| Scheme 40 Retrosynthesis of the lomaiviticin aglycon (206) by Herzon and co-workers. | ||
The monomeric diazofluorene 208 was synthesized by adaptation of the route previously developed for the synthesis of (−)-kinamycin F (3).58 The route to 208 began with silylation of 3-ethylphenol (209) to form the silyl ether 210 (>99%, Scheme 41). Birch reduction and asymmetric dihydroxylation then formed the diol 211 (61%, 91% ee). Palladium-mediated oxidation of the enoxysilane of 211 generated the α,β-unsaturated ketone 212 (92%). A mixture of diastereomeric mesityl aldehyde acetals 213 was then formed by heating of 212 in the presence of mesityl aldehyde dimethylacetal and pyridinium para-toluenesulfonate (85%, 1:1 dr).
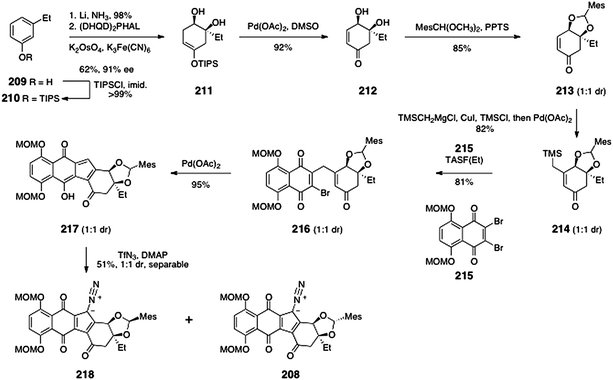 | ||
| Scheme 41 Synthesis of the diazofluorenes 218 and 208 by Herzon and co-workers. | ||
Copper-catalyzed addition of trimethylsilylmethylmagnesium chloride, trapping of the resulting enolate with chloro trimethylsilane, and reoxidation then formed the β-(trimethylsilylmethyl)-α,β-unsaturated ketone 214 (82%, 1:1 dr). TASF(Et)-mediated coupling of 214 and the dibromonaphthoquinone 215 then formed the γ-quinonylation product 216 (81%). Palladium-mediated cyclization furnished the hydroxyfulvene 217 (95%). Finally, diazo transfer to 217 generated the diazofluorenes 218 and 208 (51% combined yield), which were separated by flash-column chromatography.
In order to dimerize the monomeric diazofluorene 208, the authors first converted it to the trimethylsilyl enoxysilane 219 (Scheme 42). Treatment of the enoxysilane 219 with a variety of conventional oxidants resulted in aromatization of the substrate (see Scheme 21). Ultimately, the authors found that manganese tris(hexafluoroacetylacetonate) (220)78,79 was effective in promoting the critical dimerization reaction. Thus, treatment of 219 with 220 in benzene at 21 °C formed the desired (2R, 2′R)-dimer 207 (26–33%), along with the alternate C2-symmetric dimer 221 (12–23%). The authors found that the orientation of the mesityl protecting group was essential to obtain the desired (2R, 2′R)-isomer (207). Dimerization of the endo-mesityldiazofluorene 218 formed the undesired (2S, 2′S)-dimer exclusively (not shown). It was proposed that the mesityl group of 219 may gear the C-3 ethyl substituent over the over the Si face of the enoxysilane, forcing bond formation to occur from the desired Re face (Fig. 5).
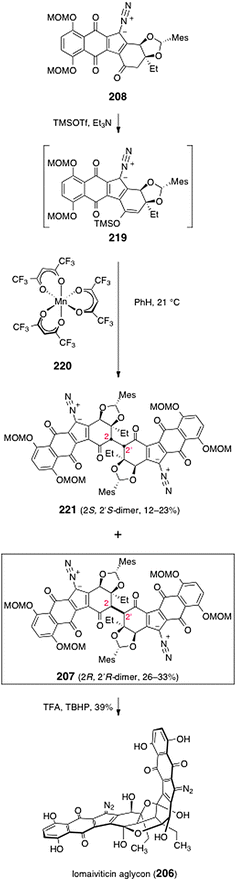 | ||
| Scheme 42 Conversion of the monomeric diazofluorene 208 to lomaiviticin aglycon (206). | ||
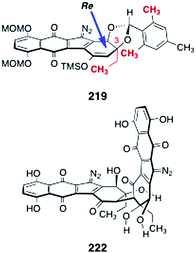 | ||
| Fig. 5 Stereochemical model for the dimerization of 219 and the structure of the open-chain isomer of lomaiviticin aglycon (222). | ||
Deprotection of 207 was effected by treatment with excess trifluoroacetic acid and tert-butylhydroperoxide in dichloromethane (39%). The authors determined that the open-chain isomer of the lomaiviticin aglycon (222, Fig. 5) was the product that was first formed in the deprotection step. The open-chain isomer 222 was observed to cyclize to lomaiviticin aglycon (206) on standing in chloroform-d or upon purification by flash-column chromatography.
6 Syntheses of the lomaiviticin carbohydrates and glycosylation studies
6.1 Introduction
The lomaiviticins contain one or two pairs of 2,6-dideoxyglycosides, β-N,N-dimethyl-pyrrolosamine (223) and α-oleandrose (224, Fig. 6). Oleandrose is a relatively common sugar that is found in a number of metabolites, such as the avermectins.80,81 In contrast, N,N-dimethyl-pyrrolosamine (223) has been found in only one other metabolite, pyrrolosporin.26,82 While several synthetic routes to both D- and L-oleandrose have been reported,83–90N,N-dimethyl-pyrrolosamine (223) had not been synthesized prior to work in the lomaiviticin area.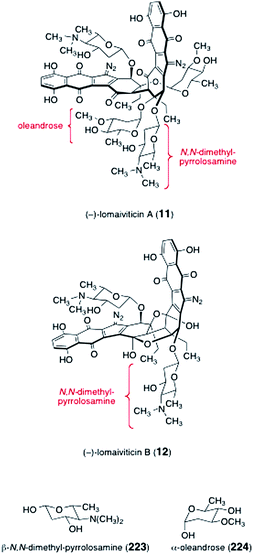 | ||
| Fig. 6 Structures of lomaiviticins A and B (11, 12, respectively), N,N-dimethyl pyrrolosamine (223), and oleandrose (224). | ||
The oleandrose residue of the lomaiviticins is α-linked. This epimer is both thermodynamically and (typically) kinetically favored in glycosylation reactions. By comparison, the N,N-dimethyl-pyrrolosamine (223) residue is β-linked. The construction of β-linked 2-deoxyglycosides is notoriously difficult and often involves installation of a removable C-2 substituent to control stereochemistry in the glycosylation event.91 However, these C-2 substituents are most often removed under reducing conditions, which are incompatible with the diazofluorene (vide infra). Consequently, the presence of this linkage in the lomaiviticins has inspired new synthetic methods, in particular from the Shair laboratory (vide infra).
6.2 Synthesis of the pyrrolosamine residue and the direct formation of β-2-deoxyglycosides by O-alkylation (Shair and co-workers)
Shair and co-workers have published two approaches to N,N-dimethyl-pyrrolosamine (223)92,93 as well as a direct method for the synthesis of β-linked 2-deoxyglycosides by O-alkylation. Shair's first approach to 223 begins with the known glycal 225 (Scheme 43).94 Treatment with trifluoroacetic anhydride followed by displacement of the resulting secondary triflate (not shown) with azide formed the azidoglycal 226 (61%, two steps). The azidoglycal 226 was then hydroacetoxylated to form a glycosyl acetate (81%, not shown). Cleavage of the acetate (dimethylamine) then formed the protected pyrrolosamine derivative 227 as a 4:1 mixture of anomers (86%).
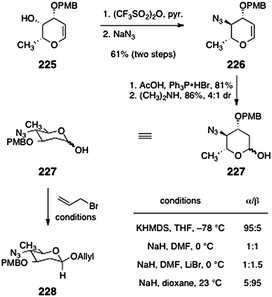 | ||
| Scheme 43 First-generation synthesis of the protected pyrrolosamine derivative 227 and optimization of the anomeric O-alkylation reaction (Shair and co-workers). | ||
In the course of these studies, the authors developed a kinetically-controlled β-selective anomeric O-alkylation.92 Thus, treatment of 227 with allyl bromide and sodium hydride in dioxane at 23 °C formed the β-linked glycoside 228 with excellent selectivity (up to 20:1). The authors showed that these conditions were amenable to a range of 2-deoxyglycosides and electrophiles. Although β-selective anomeric O-alkylation reactions had previously been reported,95–98 the studies of Shair and co-workers clearly established that the enhanced β-selectivity arises from destabilizing interactions of the β-anomeric alkoxide with the endocyclic oxygen (rather than from repulsion with the C-2 substituent; see Scheme 44).
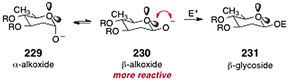 | ||
| Scheme 44 Lone-pair repulsion model to rationalize the β-selectivity in the anomeric O-alkylation reaction (Shair and co-workers). | ||
The same group also reported a second synthesis of protected pyrrolosamine from L-threonine (232, Scheme 45).93 The starting material 232 was converted to the oxazolidine 233 by treatment with pyridinium para-toluenesulfonate and pivaldehyde (71%, 15:1 dr). Deprotonation of 233 (lithium di-iso-propylamide) followed by treatment with acetic acid afforded the diastereomeric product 234 (>99%). The methyl ester function of 234 was converted to a Weinreb amide (not shown). Reaction of this amide with ethynyl Grignard afforded the alkynyl ketone 235 (68%, two steps). In the next step, diasteroselective reduction provided a propargylic alcohol (not shown) that was protected with tri-iso-propylsilyl trifluoromethanesulfonate to yield the silyl ether 236 (88%, two steps). Treatment of 236 with acetic acid cleaved the oxazolidine ring to give the aminoalcohol 237 (58%). Finally, rhodium-catalyzed cycloisomerization provided the glycal 238 (76%), which may be elaborated to N,N-dimethyl-pyrrolosamine (223).
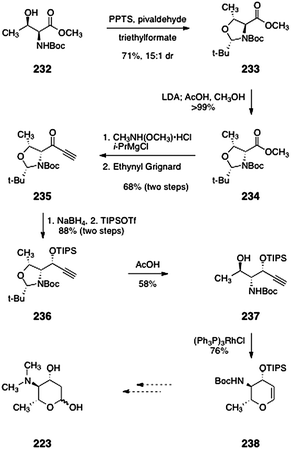 | ||
| Scheme 45 Synthesis of the glycal 238 (Shair and co-workers). | ||
6.3 Synthesis of N,N-dimethyl-pyrrolosamine and oleandrose. Incorporation into a lomaiviticin model system (Herzon and co-workers)
Herzon and co-workers developed a strategy to synthesize either the D- or L-isomers of the oleandrose and N,N-dimethyl-pyrrolosamine residues of the lomaiviticins.99 The route to either sugar begins with the known epoxide 239 or its enantiomer ent-239 (Scheme 46), which are available in one step by epoxidation of ethyl sorbate using the Shi catalyst.100 Heating of the epoxide 239 in the presence of dimethylamine formed the aminoalcohol 240 (51%). Reduction of the ester of 240 (di-iso-butylaluminum hydride, −78 °C) afforded the aldehyde 241 (>90%). The aldehyde 241 was then treated with DBU in allyl alcohol as solvent to furnish O-allyl-N,N-dimethyl-D-pyrrolosamine (242, 88%). Alternatively, opening of the epoxide ent-239 with allyl alcohol using sulphuric acid as catalyst afforded the alcohol 243 (56%). Reduction of the ester of 243 (di-iso-butylaluminum hydride, −78 °C) provided the unsaturated aldehyde 244 (89%). Finally, treatment of the aldehyde 244 with DBU in methanol provided O-allyl-L-oleandrose (245) as well as its C-3 epimer (not shown) in a 1:1 ratio (83%). Resubjection of the C-3 epimer to the reaction conditions served to regenerate the 1:1 mixture and increase material throughput.
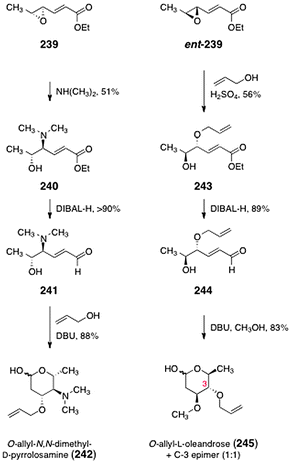 | ||
| Scheme 46 Synthesis of O-allyl-N,N-dimethyl-D-pyrrolosamine (242) and O-allyl-L-oleandrose (245) by Herzon and co-workers. | ||
With the carbohydrates of the lomaiviticins in hand, Herzon and co-workers studied their regio- and stereoselective incorporation into the model diol 212 (Scheme 47). The authors found that the pyrrolosamine derivative 242 was best activated as its corresponding glycosylphosphinate 246 (47%, α-anomer).101–103 Treatment of mixtures of the diol 212 and the anomerically-pure α-phosphinate 246 with borontrifluoride etherate furnished a mixture of α- and β-glycosides 248 (57%, α:β = 2:3). For the second glycosylation, the protected oleandrose derivative 245 was activated with diethylamino sulfurtrifluoride (DAST, 81%)104 to afford the glycosyl fluoride 247. Treatment of β-248 with the fluoride 247 in the presence of borontrifluoride etherate then formed the bis(glycoside) 249 in 33% yield.
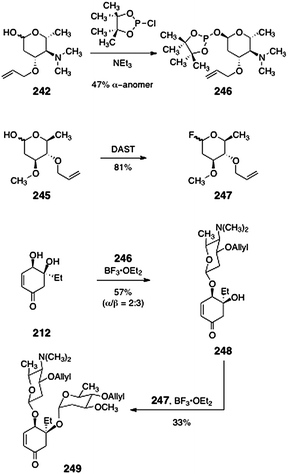 | ||
| Scheme 47 Synthesis of the lomaiviticin cyclohexenone bis(glycoside) by sequential glycosylation of the diol 212. | ||
7 Syntheses of metabolites related to the kinamycins and lomaiviticins
7.1 Introduction
A number of kinamycin biosynthetic precursors or shunt metabolites in the biosynthetic pathway have been isolated from the producing strain of the kinamycins. In this section, we present the syntheses of these metabolites.7.2 Prekinamycin (original and revised structures; Hauser, Echavarren, Birman, and Ishikawa groups)
As discussed in Section 2, prekinamycin (18) was isolated from the producing strain of the kinamycins by Gould and co-workers.8 The structure of prekinamycin was originally proposed to be that depicted as 17 and was subsequently revised to 18 (Fig. 7).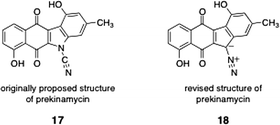 | ||
| Fig. 7 Originally proposed structure of prekinamycin (17) and revised (correct) structure (18). | ||
In 1996, Hauser and Zhou reported a synthesis of prekinamycin (18, Scheme 48).105 The authors began with the readily-available phthalide sulfone 250 and indenone 251. Treatment of a mixture of 250 and 251 with lithium tert-butoxide resulted in efficient annulation to afford the tetracyclic ketone 252 (73%). The ketone 252 was then demethylated by treatment with boron tribromide. Hydrazone formation (hydrazine) and oxidation (silver carbonate) afforded prekinamycin (18, yield not specified). Mal and Hazra utilized Hauser's strategy to synthesize related, but simpler, tetracyclic products.106
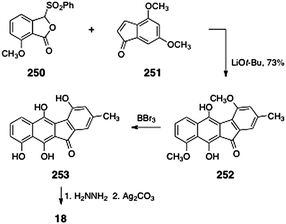 | ||
| Scheme 48 Synthesis of prekinamycin (18) by Hauser and co-workers. | ||
The cyanocarbazole structure originally proposed for prekinamycin (17) was prepared by Echavarren and co-workers.23 As discussed above, this synthetic work was instrumental in establishing the diazofluorene structure of the kinamycins and lomaiviticins (see Section 2.1). Echavarren's synthesis of 17 began with Stille coupling between the bromojuglone derivative 254 and the arylstannane 255 (99%, Scheme 49). Oxidation with basic tert-butyl hydrogen peroxide provided the hydroxynaphthoquinone 257 (87%). Thermolysis of the hydroxynaphthoquinone 257 induced dehydrative cyclization, forming the expected product 259 (20%) as well as the rearranged product 258 (20%). The desired product 259 was cyanated to form the cyanocarbazole 260 (30–76%) Demethylation (boron tribromide) afforded the originally proposed structure of prekinamycin (17, 90–100%).
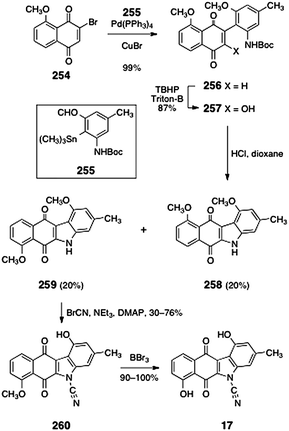 | ||
| Scheme 49 Synthesis of the originally-proposed structure of prekinamycin (17) by Echavarren and co-workers. | ||
In 2007, Birman and co-workers reported a highly efficient synthesis of the revised structure of prekinamycin (18, Scheme 50).107 Their synthesis began with base-mediated double acylation of the indanone 262 with the phthalate ester 261 (67–81%). Under these conditions, it was proposed that the dianion of 262 is generated. This dianion undergoes regioselective attack at the more electrophilic ester of the phthalate (see structure 263). In situ Dieckmann condensation then forms the observed tetracyclic product 264. Condensation of 264 with methanesulfonylhydrazine formed the hydrazone 265 (99%). Demethylation (boron tribromide) afforded the tetraol 266. Treatment of 266 with triethylamine under an atmosphere of air resulted in spontaneous oxidation to afford prekinamycin (18, 85%, two steps).
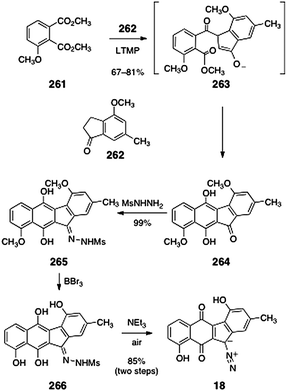 | ||
| Scheme 50 Synthesis of prekinamycin (18) by Birman and co-workers. | ||
In 2011, Ishikawa and coworkers reported a synthesis of prekinamycin (18, Scheme 51).108 Their synthesis began with a Suzuki coupling between the aryl boronic acid 267 and the bromide 268108 (97%). Oxidation of the coupling product 269 with basic hydrogen peroxide formed the carboxylic acid 270 (80%). Conversion to the acyl chloride (oxalyl chloride) and intramolecular Friedel–Crafts reaction gave the tetracyclic ketone 271 (81%, two steps). Cleavage of the methoxymethyl and methyl protecting groups (boron tribromide) then provided the tetraol 272. Treatment of the unpurified tetraol with para-toluenesulfonylhydrazine and oxidation with silver carbonate then afforded prekinamycin (18, 47%, three steps).
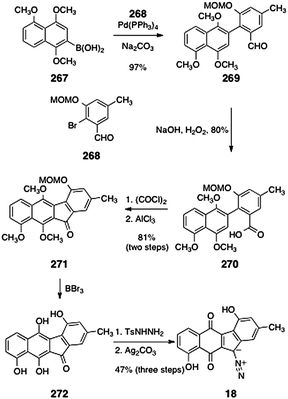 | ||
| Scheme 51 Synthesis of prekinamycin (18) by Ishikawa and co-workers. | ||
7.3 Isoprekinamycin (Dmitrienko and co-workers)
Dmitrienko and co-workers reported the total synthesis of isoprekinamycin (281, Scheme 52),109 a diazobenzo[a]fluorene natural product that was isolated from the producing strain of the kinamycins.8,110 The synthesis began with the readily-available alcohol 273, which was converted to the nitrile 274 by a two-step sequence (69%). Palladium-mediated borylation afforded the arylboronate ester 275 (82%). Suzuki coupling of the arylboronate ester 275 with the bromoindenone 276 afforded the α-arylketone 277 (85%). The α-arylketone 277 was reduced with a mixture of n-butyllithium and di-iso-butylaluminumhydride,111 to form the putative aluminium alkoxide intermediate 278. Addition of lithium di-iso-propylamide to the reaction mixture promoted cyclization to afford the tetracycle 279. The tetracycle 279 was then O-methylated (potassium carbonate, methyl iodide) and the nitrile function was hydrolyzed (hydrogen peroxide, potassium carbonate, methyl sulfoxide). Under these conditions, the secondary alcohol was also oxidized, providing the amidoketone 280 in 51% yield from 277. Finally, Hoffmann rearrangement, cleavage of the resulting carbamate, diazotization, and demethylation, afforded isoprekinamycin (281, 46%, four steps).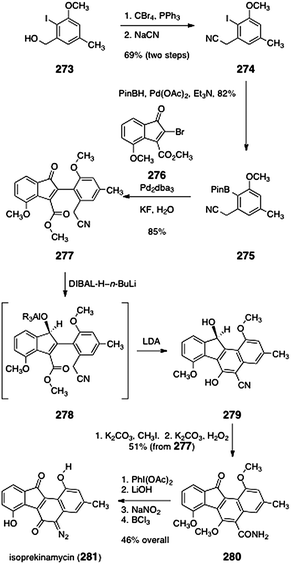 | ||
| Scheme 52 Synthesis of isoprekinamycin (281) by Dmitrienko and co-workers. | ||
7.4 Kinobscurinone (Snieckus and co-workers)
A formal total synthesis of kinobscurinone (21, Scheme 3) was reported by Snieckus and co-workers.112 Their synthesis began with Suzuki coupling of the aryl bromide 282 and the boronic acid 283 (95%, Scheme 53). The coupling product 284 was then treated with excess s-butyllithium to generate an ortho-metalated aryl carbamate (not shown). Trapping with chlorotrimethylsilane formed the aryl silane 285 (97%). Treatment of 285 with excess lithium di-iso-propylamide induced arene metalation–carbamoyl transfer to form the aryl amide 286 (62%). Methylation of 286 formed the tetramethyl ether 287 (96%). Treatment of 287 with excess lithium di-iso-propylamide generated the tetracycle 288 (78%). Protodesilylation (trifluoroacetic acid, reflux) produced tetramethylkinobscurinone (289, quant.). Tetramethylkinobscurinone (289) had previously been transformed to kinobscurinone (21) by Gould and co-workers.27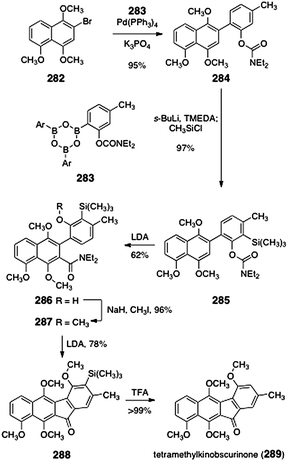 | ||
| Scheme 53 Synthesis of tetramethylkinobscurinone (289) by Snieckus and co-workers. | ||
7.5 Stealthins (Gould and Kamikawa groups)
Stealthins A–C (290, 291, and 22, respectively, Fig. 8) were isolated from the fermentation broth of various strains of Streptomyces.28,113 Owing to the presence of a stable free radical valence tautomer, these metabolites are NMR-silent and exhibit potent radical scavenging activity.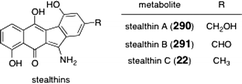 | ||
| Fig. 8 Structures of the stealthins A, B, and C (290, 291, and 22, respectively). | ||
Gould reported the synthesis of stealthin C (22) from tetramethylkinobscurinone (289, Scheme 54).28 Treatment of tetramethylkinobscurinone (289) with hydroxylamine formed the oxime 292 (99%). Cleavage of the methyl ethers afforded the quinone 293 (69%). Finally, dithionite reduction provided stealthin C (22, 80%).
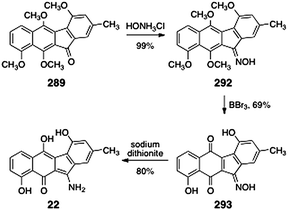 | ||
| Scheme 54 Synthesis of stealthin C (22) from tetramethylkinobscurinone (289) by Gould and co-workers. | ||
Kamikawa and Koyama reported the synthesis of stealthin C dimethyl ether (301, Scheme 55).114 The synthesis began with a Suzuki coupling of the arylboronic acid 294 and the aryl bromide 295 (100%). Oxidation of the aldehyde function of the coupling product 296 afforded the carboxylic acid 297 (91%). Activation of the carboxylic acid (oxalyl chloride) formed an acid chloride (not shown), that on treatment with titanium chloride underwent efficient Friedel–Crafts ring closure, to afford the tetracyclic ketone 298 (79%). Condensation of 298 with O-benzyl hydroxylamine generated an oxime ether (77%, not shown). Oxidative demethylation using ceric ammonium nitrate and pyridine 2,6-dicarboxylic acid N-oxide (299) afforded the naphthoquinone 300 (77%). Finally, reduction of 300 (zinc, acetic acid) afforded stealthin C dimethyl ether (301, 80%).
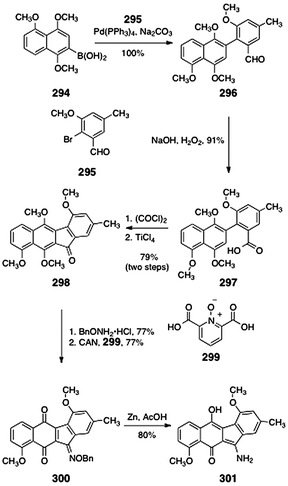 | ||
| Scheme 55 Synthesis of stealthin C dimethyl ether (301) by Kamikawa and Koyama. | ||
8 Chemical biological studies
8.1 Introduction
The kinamycins and lomaiviticins are potent anticancer and antimicrobial agents. (−)-Kinamycin C (3) exhibited a <GI50> = 340 nM against the NCI 60 cell line panel.14 (−)-Lomaiviticin A (11) exhibited IC50 values in the low nanomolar range, with activities as low as 10 pM against ovarian cell lines.12 Moreover, the toxicity profiles of 3 and 11 are unique, which is indicative of a novel mechanism of action. Various kinamycins and lomaiviticins also inhibit the growth of Gram-positive and Gram-negative bacteria at low micromolar–nanomolar concentrations.12Diazo and diazonium-based DNA cleaving agents, and their mode of action, have been reviewed.18 Herein, we will discuss studies that relate directly to the kinamycins and lomaiviticins.
8.2 Chemical biological studies of the kinamycins and lomaiviticins (Melander, Hasinoff, and Dimitrienko groups)
Melander and co-workers have shown that the kinamycins cleave dsDNA in the presence of a reducing co-factor.115,116 This dsDNA cleavage appears to be non-sequence specific, suggesting a free radical mechanism may be operative. Experiments employed dithiothrietol (DTT) or glutathione (GSH) at intracellularly-relevant concentrations. The authors speculated that reduction of the quinone to form a hydroquinone, such as 302, may occur (Scheme 56). Reduction of the quinone may trigger loss of the diazo function to form a reactive ortho-quinone methide (303). Alternatively, the hydroquinone 302 may form an adduct with an exogenous nucleophile (see structure 304). The adduct (304) may undergo decomposition to form alkyl-centered radicals, which would be expected to damage DNA. These hypotheses are similar to those put forth independently by the Hasinoff–Dimitrienko and Feldman research groups (vide infra). It is quite possible that the observed cytotoxic effects of the kinamycins arise from a combination of these reaction pathways.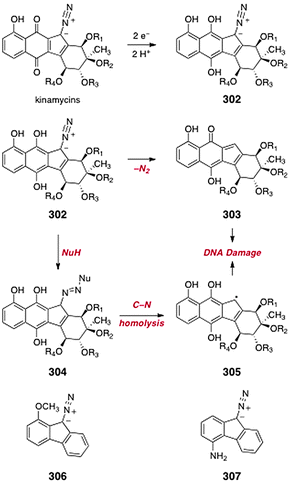 | ||
| Scheme 56 Proposed reaction pathways for the kinamycins under reducing conditions (Melander and co-workers). | ||
The Melander group has also shown that simple diazofluorenes, such as 306 and 307, are able to cleave dsDNA,117 suggesting the diazofluorene as a general DNA-cleaving functional group. The substitution pattern about the diazofluorenes had a measurable effect on their cleavage ability. 1-Methoxydiazofluorene (306) exhibited DNA cleaving abilities similar to kinamycin D (4), whereas 4-aminodiazofluorene (307) was found to cleave DNA even in the absence of a reducing agent.
Hasinoff and Dmitrienko have suggested that the cytotoxicity of the kinamycins may be due to generation of reactive oxygen species (ROS) in vivo.118 Consistent with Melander's studies, these authors observed reaction between kinamycin F (6) and GSH. Electron paramagnetic resonance studies showed that the reaction of kinamycin F (6) with GSH formed a semiquinone free radical. By modulating the intracellular concentration of GSH, the authors were able to show that the toxicity of kinamycin F (6) correlated linearly with [GSH], implicating this reaction pathway in the cytotoxic effects of kinamycin F (6). The reagent 2-oxo-4-thiazolidinecarboxylic acid was used to increase cellular GSH levels in K562 cells. Buthionine sulfoximine was used to decrease GSH levels in the same cell line. The authors also showed that kinamycin F (6) weakly bound DNA. Using an ethidium bromide displacement assay, the authors determined Kapp to be 9 × 104 M−1. This is ca. 300-fold weaker than doxorubicin. In vitro studies revealed that kinamycin F (6) induced small amounts of DNA cleavage in the absence of a reducing co-factor. In the presence of GSH, kinamycin F (6) efficiently cleaved DNA. This cleavage activity decreased on addition of deferoxamine, dimethyl sulfoxide, and catalase, suggesting DNA damage occurs in an iron-, hydrogen peroxide-, and hydroxyl radical-dependent fashion.
Additional studies using synchronized K562 cells showed an accumulation of cells in the S phase, and delayed entry into the G2/M phase on treatment with kinamycin F (6).118 Kinamycin F (6) activated the caspase-3/7 pathway, leading to an increase in apoptosis. The authors show that the production of cyclin D3 was diminished in cells treated with kinamycin F (6), although the protein itself was stable in the presence of 6. It was shown that 6 decreased the level of cyclin D3 mRNA, but had little interaction with the DNA or mRNA itself. In light of the poor DNA and mRNA binding and high concentrations of kinamycins required for DNA damage, the authors speculated that the kinamycins may target a protein in the cyclin D3 production pathway.
Little is known about the mechanism of action of the lomaiviticins, most likely due to difficulties associated with their fermentation. In the isolation report, lomaiviticin A (11) was reported to cleave dsDNA under reducing conditions.12 Further studies are required to fully evaluate this reactivity.
8.3 In vitro reactivity studies (Jebaratnam, Dmitrienko, Feldman, Skibo, and Moore groups)
The in vitro and tissue culture reactivity of simple diazofluorenes has been studied by several groups. In 1995, Arya and Jebaratnam reported that DNA was efficiently cleaved when incubated in the presence of the diazofluorene 308 and cupric acetate (Scheme 57).119 Control experiments indicated that both reagents were required for cleavage. The authors proposed that the diazofluorene 308 may undergo oxidation to the free radical 309 and that the latter might be the active DNA-cleaving agent. It was suggested that the quinone function of the kinamycins may act as an internal oxidant toward the diazo group, triggering the formation of a radical similar to 309. However, other studies seem to suggest that the kinamycins and lomaiviticins are reductively activated. | ||
| Scheme 57 Proposed oxidation of 308 to form a reactive free radical intermediate (Arya and Jebaratnam). | ||
Dmitrienko and co-workers studied the in vitro reactivity of isoprekinamycin (281, Scheme 58).120 It was shown that isoprekinamycin (281) underwent addition of β-napthol to form the alkylation product 310 (29%), as well as the reduced product 311 (61%). The authors proposed that a hydrogen-bonding interaction between the quinone and the adjacent phenol (see structure 281) may increase the electrophilicity of the α-diazoketone function of isoprekinamycin (281). The related synthetic intermediate 312, which lacks this interaction, was unreactive under otherwise identical conditions. The authors suggested that the kinamycins and lomaiviticins may exert their cytotoxic effects by undergoing addition of a biological nucleophile to the diazo function, or alternatively, by formation of vinyl radicals via loss of dinitrogen from nucleophilic addition products (e.g., 310).
 | ||
| Scheme 58 Reaction of isoprekinamycin (281) with β-naphthol under basic conditions (Dmitrienko and co-workers). | ||
Feldman and Eastman have provided convincing evidence that the kinamycins form reactive vinyl radical and ortho-quinone methide intermediates under reducing conditions (Scheme 59).121 The authors demonstrated that the alkenyl radical 314 is generated when the model substrate dimethyl prekinamycin (313) is exposed to reductants, such as tri-n-butyltin hydride. Products that may arise from addition of this radical (314) to aromatic solvents (benzene, anisole, and benzonitrile) were isolated. The ortho-quinone methide 315 was also formed, presumably by hydrogen atom abstraction by 314. Alkylation products derived from nucleophilic addition to 315 were isolated. Accordingly, it was proposed that the kinamycins and lomaiviticins may exert their cytotoxic effects by generation of free radical or electrophilic ortho-quinone methide intermediates, following reductive activation. In a separate study, Skibo and co-workers examined the reduction of a series of prekinamycin analogs (Scheme 60).122 It was established that catalytic hydrogenation of prekinamycin derivatives (316) forms ortho-quinone methide intermediates, such as 317. Interestingly, these ortho-quinone methides were found to be in equilibrium with their keto tautomers (318), and the position of equilibrium was shown to be substituent-dependent. Prekinamycin analogs that exhibited the most stable ortho-quinone methide tautomers were found to be the most cytotoxic, suggesting these intermediates may be involved in the biological effects of the kinamycins and lomaiviticins.
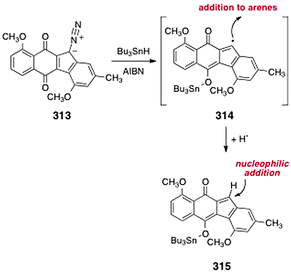 | ||
| Scheme 59 Reaction pathway for dimethylprekinamycin (313) established by Feldman and Eastman. | ||
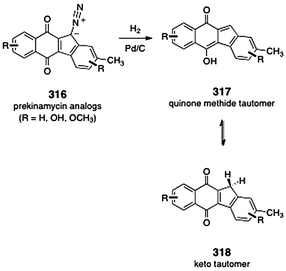 | ||
| Scheme 60 Diazofluorene reduction and tautomeric equilibria observed by Skibo and co-workers. | ||
Finally, in 1977, Moore proposed a novel mode of reactivity for kinamycin C (3, Scheme 61).123 Moore originally formulated this postulate using the cyanocarbazole structure of the kinamycins. It is shown here for the corrected structure of kinamycin C (3). Reduction of kinamycin C (3) to the hydroquinone, followed by elimination of acetic acid, was postulated to form the vinylogous ortho-quinone methide 320. This species may then eliminate a second equivalent of acetic acid, to form the double quinone methide 321. It was proposed that both 320 and 321 may incorporate biological nucleophiles, leading to cell death.
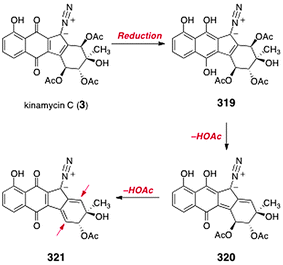 | ||
| Scheme 61 Moore's proposal for the bioreductive activation of kinamycin C (3). | ||
9 Related metabolites
9.1 Introduction
Many interesting metabolites related to the kinamycins and lomaiviticins have been isolated. A recent review of diazo-containing natural products, by Nawrat and Moody,17 contains a comprehensive overview of naturally-occurring diazo compounds. A selection of related metabolites, which lack a diazo group, are discussed below.9.2 Momofulvenones
Momofulvenones A (322) and B (323) were isolated as yellow salts (counter ion not specified) from the broth of Streptomyces diastatochromogenes by Zeeck and co-workers (Fig. 9).124 The isolates displayed similar UV spectra under neutral or basic conditions, which lent support to their proposed anionic structures. The structures of the momofulvenones were ultimately corroborated by mass spectroscopy of chemical derivatives. Labeling studies established that the biosyntheses of the kinamycins and momofulvenones follow similar pathways. The momfulvenones are not reported to possess any biological activity, perhaps an indication of the importance of the diazofunction in the kinamycins and lomaiviticins.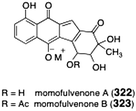 | ||
| Fig. 9 Structures of momfulvenones A (322) and B (323). | ||
9.3 Seongomycin
Seongomycin (324) was observed as a minor constituents of Streptomyces murayamaensis, and later isolated as the major metabolite in Streptomyces lividans transformed by the kinamycin gene cluster (Fig. 10).125 Although seongomycin (324) was not observed from the wild type strain, it contained the characteristic 6,6,5,6 tetracyclic scaffold of the diazofluorene natural products, and appears to be a simple adduct of N-acetyl cysteine. The side chain was established by NMR and FAB-MS analysis. The authors subsequently confirmed this structure via the biosynthetic incorporation of [1,2-13C2]acetate units. Incorporation of a cysteine unit is rare in polyketide metabolites from Streptomyces.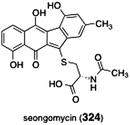 | ||
| Fig. 10 Structure of seongomycin (324). | ||
10 Conclusion and outlook
We have attempted in this review to compile the literature related to the kinamycins and lomaiviticins in one location to aid researchers in this exciting area. Although much progress has been made, several significant issues remain unresolved. First and foremost, the lomaiviticins have not yet succumbed to total synthesis, and their absolute stereochemistry remains unknown. Second, although a mounting body of evidence suggests the kinamycins and lomaiviticins are reductively-activated, a clear understanding of the molecular mechanisms underlying the cytotoxic effects of these compounds has not yet been obtained. Additionally, the biological target of these molecules has not been unequivocally established: although they have demonstrated DNA-cleaving abilities, the studies of Hasinoff and Dmitrienko seem to suggest one or more protein targets may exist. An intriguing possibility is that the molecules may be multipotent, that is, they may exert their cytotoxic effects through multiple mechanisms of action. Finally, the structural features of the lomaiviticins that impart such high levels of cytotoxicity to these metabolites have not been identified. Many of these issues are uniquely addressable by chemical synthesis, and future synthetic and chemical biological studies will bring resolution to these important questions.
Note added in proof
The synthesis of the epoxykinamycin FL-120B́ (325, Fig. 11)10,11 was recently reported by Scully and Porco.126
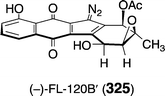 | ||
| Fig. 11 The structure of the epoxykinamycin derivative (−)-FL-120B′ (325). | ||
11 References and notes
- The producing strain of the lomaiviticins was originally classified as Micromonospora. This strain has subsequently been reclassified as Salinispora. Personal communication from Jeffrey Janso, 2011.
- S. Ito, T. Matsuya, S. Ōmura, M. Otani and A. Nakagawa, J. Antibiot., 1970, 23, 315–317 CAS
.
- T. Hata, S. Ōmura, Y. Iwai, A. Nakagawa and M. Otani, J. Antibiot., 1971, 24, 353–359 CAS
- S. Ōmura, A. Nakagawa, H. Yamada, T. Hata, A. Furusaki and T. Watanabe, Chem. Pharm. Bull., 1971, 19, 2428–2430 Search PubMed
- S. Ōmura, A. Nakagawa, H. Yamada, T. Hata and A. Furusaki, Chem. Pharm. Bull., 1973, 21, 931–940 Search PubMed
- M. C. Cone, P. J. Seaton, K. A. Halley and S. J. Gould, J. Antibiot., 1989, 42, 179–188 CAS
- K. Isshiki, T. Sawa, H. Naganawa, N. Matsuda, S. Hattori, M. Hamada, T. Takeuchi, M. Oosono, M. Ishizuka, Z. Yang, B. Zhu and W. Xu, J. Antibiot., 1989, 42, 467–469 CAS
- P. J. Seaton and S. J. Gould, J. Antibiot., 1989, 42, 189–197 CAS
- T. A. Smitka, R. Bonjouklian, T. J. Perun, A. H. Hunt, R. S. Foster, J. S. Mynderse and R. C. Yao, J. Antibiot., 1992, 45, 581–583 CAS
- H.-C. Lin, S.-C. Chang, N.-L. Wang and L.-R. Chang, J. Antibiot., 1994, 47, 675–680 CAS
- H.-C. Lin, S.-C. Chang, N.-L. Wang and L.-R. Chang, J. Antibiot., 1994, 47, 681–687 Search PubMed
- H. He, W. D. Ding, V. S. Bernan, A. D. Richardson, C. M. Ireland, M. Greenstein, G. A. Ellestad and G. T. Carter, J. Am. Chem. Soc., 2001, 123, 5362–5363 CrossRef CAS
- W. G. Overend, C. W. Rees and J. S. Sequeira, J. Chem. Soc., 1962, 3429–3440 RSC
- R. H. Shoemaker, Nat. Rev. Cancer, 2006, 6, 813–823 CrossRef CAS
- S. J. Gould, Chem. Rev., 1997, 97, 2499–2510 CrossRef CAS
- J. Marco-Contelles and M. T. Molina, Curr. Org. Chem., 2003, 7, 1433–1442 CrossRef CAS
- C. C. Nawrat and C. J. Moody, Nat. Prod. Rep., 2011, 28, 1426–1444 RSC
- D. P. Arya, Top. Heterocycl. Chem., 2006, 2, 129–152 CrossRef CAS
- A. Furusaki, M. Matsui, T. Watanabe, S. Ōmura, A. Nakagawa and T. Hata, Isr. J. Chem., 1972, 10, 173–187 CAS
- K. Ajisaka, H. Takeshima and S. Ōmura, J. Chem. Soc., Chem. Commun., 1976, 571 RSC
- P. J. Seaton and S. J. Gould, J. Am. Chem. Soc., 1988, 110, 5912–5914 CrossRef CAS
- G. I. Dmitrienko, K. E. Nielsen, C. Steingart, S. M. Ngai, J. M. Willson and G. Weeratunga, Tetrahedron Lett., 1990, 31, 3681–3684 CrossRef CAS
- A. M. Echavarren, N. Tamayo and M. C. Paredes, Tetrahedron Lett., 1993, 34, 4713–4716 CrossRef CAS
- S. J. Gould, N. Tamayo, C. R. Melville and M. C. Cone, J. Am. Chem. Soc., 1994, 116, 2207–2208 CrossRef CAS
- S. Mithani, G. Weeratunga, N. J. Taylor and G. I. Dmitrienko, J. Am. Chem. Soc., 1994, 116, 2209–2210 CrossRef CAS
- D. R. Schroeder, K. L. Colson, S. E. Klohr, M. S. Lee, J. A. Matson, L. S. Brinen and J. Clardy, J. Antibiot., 1996, 49, 865–872 CAS
- S. J. Gould and C. R. Melville, Bioorg. Med. Chem. Lett., 1995, 5, 51–54 CrossRef CAS
- S. J. Gould, C. R. Melville, M. C. Cone, J. Chen and J. R. Carney, J. Org. Chem., 1997, 62, 320–324 CrossRef CAS
- N. Chen, M. B. Carriere, R. S. Laufer, N. J. Taylor and G. I. Dmitrienko, Org. Lett., 2008, 10, 381–384 CrossRef CAS
- S. J. Gould, T. O'Hare, P. Seaton, J. Soodsma and Z. Tang, Bioorg. Med. Chem., 1996, 4, 987–994 CrossRef CAS
- X. Lei and J. A. Porco, J. Am. Chem. Soc., 2006, 128, 14790–14791 CrossRef CAS
- For a review, see: S. P. Roche and J. A. Porco, Angew. Chem., Int. Ed., 2011, 50, 4068–4093 CrossRef CAS
- Y. Hu, C. Li, B. A. Kulkarni, G. Strobel, E. Lobkovsky, R. M. Torczynski and J. A. Porco, Org. Lett., 2001, 3, 1649–1652 CrossRef CAS
- V. K. Aggarwal, A. Mereu, G. J. Tarver and R. McCague, J. Org. Chem., 1998, 63, 7183–7189 CrossRef CAS
- C. L. Elston, R. F. W. Jackson, S. J. F. MacDonald and P. J. Murray, Angew. Chem., Int. Ed. Engl., 1997, 36, 410–412 CrossRef CAS
- C. Li, E. A. Pace, M.-C. Liang, E. Lobkovsky, T. D. Gilmore and J. A. Porco, J. Am. Chem. Soc., 2001, 123, 11308–11309 CrossRef CAS
- C. Li, R. P. Johnson and J. A. Porco, J. Am. Chem. Soc., 2003, 125, 5095–5106 CrossRef CAS
- M. E. Jung and J. A. Hagenah, J. Org. Chem., 1983, 48, 5359–5361 CrossRef CAS
- H. Azizian, C. Eaborn and A. Pidcock, J. Organomet. Chem., 1981, 215, 49–58 CrossRef CAS
- M. Caron and K. B. Sharpless, J. Org. Chem., 1985, 50, 1557–1560 CrossRef CAS
- M. E. Furrow and A. G. Myers, J. Am. Chem. Soc., 2004, 126, 5436–5445 CrossRef CAS
- M. E. Furrow and A. G. Myers, J. Am. Chem. Soc., 2004, 126, 12222–12223 CrossRef CAS
- Y. Kitani, A. Morita, T. Kumamoto and T. Ishikawa, Helv. Chim. Acta, 2002, 85, 1186–1195 CrossRef CAS
- T. Kumamoto, Y. Kitani, H. Tsuchiya, K. Yamaguchi, H. Seki and T. Ishikawa, Tetrahedron, 2007, 63, 5189–5199 CrossRef CAS
- S. W. Heinzman and J. R. Grunwell, Tetrahedron Lett., 1980, 21, 4305–4308 CrossRef CAS
- K. C. Nicolaou, T. Montagnon and P. S. Baran, Angew. Chem., Int. Ed., 2002, 41, 993–996 CrossRef CAS
- T. J. Donohoe, K. Blades, P. R. Moore, M. J. Waring, J. J. G. Winter, M. Helliwell, N. J. Newcombe and G. Stemp, J. Org. Chem., 2002, 67, 7946–7956 CrossRef CAS
- D. A. Evans, K. T. Chapman and E. M. Carreira, J. Am. Chem. Soc., 1988, 110, 3560–3578 CrossRef CAS
- P. Jacob, P. S. Callery, A. T. Shulgin and N. Castagnoli, J. Org. Chem., 1976, 41, 3627–3629 CrossRef CAS
- K. C. Nicolaou, H. Li, A. L. Nold, D. Pappo and A. Lenzen, J. Am. Chem. Soc., 2007, 129, 10356–10357 CrossRef CAS
- G. Giuffredi, C. Bobbio and V.r. Gouverneur, J. Org. Chem., 2006, 71, 5361–5364 CrossRef CAS
- C. R. Johnson, J. P. Adams, M. P. Braun, C. B. W. Senanayake, P. M. Wovkulich and M. R. Uskokovic, Tetrahedron Lett., 1992, 33, 917–918 CrossRef CAS
- M. G. Banwell, B. D. Kelly, O. J. Kokas and D. W. Lupton, Org. Lett., 2003, 5, 2497–2500 CrossRef CAS
- M. S. Kerr, J. Read de Alaniz and T. Rovis, J. Am. Chem. Soc., 2002, 124, 10298–10299 CrossRef CAS
- M. S. Kerr, J. Read de Alaniz and T. Rovis, J. Org. Chem., 2005, 70, 5725–5728 CrossRef CAS
- G. A. Molander and G. Hahn, J. Org. Chem., 1986, 51, 1135–1138 CrossRef CAS
- For a review, see: G. A. Molander, Chem. Rev., 1992, 92, 29–68 CrossRef CAS
- C. M. Woo, L. Lu, S. L. Gholap, D. R. Smith and S. B. Herzon, J. Am. Chem. Soc., 2010, 132, 2540–2541 CrossRef CAS
- H. C. Kolb, M. S. VanNieuwenhze and K. B. Sharpless, Chem. Rev., 1994, 94, 2483–2547 CrossRef CAS
- H. J. Reich, I. L. Reich and J. M. Renga, J. Am. Chem. Soc., 1973, 95, 5813–5815 CrossRef CAS
- K. B. Sharpless, R. F. Launer and A. Y. Teranishi, J. Am. Chem. Soc., 1973, 95, 6137–6139 CrossRef CAS
- A. B. Charette, R. P. Wurz and T. Ollevier, J. Org. Chem., 2000, 65, 9252–9254 CrossRef CAS
- W.v. E. Doering and C. H. DePuy, J. Am. Chem. Soc., 1953, 75, 5955–5957 CrossRef CAS
- K. C. Nicolaou, R. M. Denton, A. Lenzen, D. J. Edmonds, A. Li, R. R. Milburn and S. T. Harrison, Angew. Chem., Int. Ed., 2006, 45, 2076–2081 CrossRef CAS
- G.-q. Lin and R. Hong, J. Org. Chem., 2001, 66, 2877–2880 CrossRef CAS
- T. Imamoto, N. Takiyama, K. Nakamura, T. Hatajima and Y. Kamiya, J. Am. Chem. Soc., 1989, 111, 4392–4398 CrossRef CAS
- W. Zhang, A. Baranczak and G. A. Sulikowski, Org. Lett., 2008, 10, 1939–1941 CrossRef CAS
- J. E. Audia, L. Boisvert, A. D. Patten, A. Villalobos and S. J. Danishefsky, J. Org. Chem., 1989, 54, 3738–3740 CrossRef CAS
- E. S. Krygowski, K. Murphy-Benenato and M. D. Shair, Angew. Chem., Int. Ed., 2008, 47, 1680–1684 CrossRef CAS
- H. G. Lee, J. Y. Ahn, A. S. Lee and M. D. Shair, Chem.–Eur. J., 2010, 16, 13058–13062 CrossRef CAS
- H. H. Wasserman and G. M. Lee, Tetrahedron Lett., 1994, 35, 9783–9786 CrossRef CAS
- D. A. Evans, K. T. Chapman and J. Bisaha, J. Am. Chem. Soc., 1988, 110, 1238–1256 CrossRef CAS
- D. A. Evans, J. S. Tedrow, J. T. Shaw and C. W. Downey, J. Am. Chem. Soc., 2002, 124, 392–393 CrossRef CAS
- G. A. Kraus and H. Sugimoto, Tetrahedron Lett., 1978, 19, 2263–2266 CrossRef
- F. M. Hauser and R. P. Rhee, J. Org. Chem., 1978, 43, 178–180 CrossRef CAS
- K. C. Nicolaou, A. L. Nold and H. Li, Angew. Chem., Int. Ed., 2009, 48, 5860 CrossRef CAS
- S. B. Herzon, L. Lu, C. M. Woo and S. L. Gholap, J. Am. Chem. Soc., 2011, 133, 7260–7263 CrossRef CAS
- S. Evans, A. Hamnett, A. F. Orchard and D. R. Lloyd, Faraday Discuss. Chem. Soc., 1972, 54, 227–250 RSC
- J. R. Bryant, J. E. Taves and J. M. Mayer, Inorg. Chem., 2002, 41, 2769–2776 CrossRef CAS
- R. W. Burg, B. M. Miller, E. E. Baker, J. Birnbaum, S. A. Currie, R. Hartman, Y.-L. Kong, R. L. Monaghan, G. Olson, I. Putter, J. B. Tunac, H. Wallick, E. O. Stapley, R. Oiwa and S. Ōmura, Antimicrob. Agents Chemother., 1979, 15, 361–367 CAS
- T. W. Miller, L. Chaiet, D. J. Cole, L. J. Cole, J. E. Flor, R. T. Goegelman, V. P. Gullo, H. Joshua, A. J. Kempf, W. R. Krellwitz, R. L. Monaghan, R. E. Ormond, K. E. Wilson, G. Albers-Schonberg and I. Putter, Antimicrob. Agents Chemother., 1979, 15, 368–371 CAS
- K. S. Lam, G. A. Hesler, D. R. Gustavson, R. L. Berry, K. Tomita, J. L. MacBeth, J. Ross, D. Miller and S. Forenza, J. Antibiot., 1996, 49, 860–864 CAS
- F. Blindenbacher and T. Reichstein, Helv. Chim. Acta, 1948, 31, 2061–2064 CrossRef CAS
- G. Berti, G. Catelani, F. Colonna and L. Monti, Tetrahedron, 1982, 38, 3067–3072 CrossRef CAS
- P. G. M. Wuts and S. S. Bigelow, J. Org. Chem., 1983, 48, 3489–3493 CrossRef CAS
- D. Ravi, V. R. Kulkarni and H. B. Mereyala, Tetrahedron Lett., 1989, 30, 4287–4290 CrossRef CAS
- R. L. Tolman and L. H. Peterson, Carbohydr. Res., 1989, 189, 113–122 CrossRef CAS
- M. J. Ford and S. V. Ley, Synlett, 1990, 771–772 CrossRef CAS
- M. W. Bredenkamp, C. W. Holzapfel and F. Toerien, Synth. Commun., 1992, 22, 2459–2477 CrossRef CAS
- X. Y. Zhao, M. Ono, H. Akita and Y. M. Chi, Chin. Chem. Lett., 2006, 17, 730–732 CAS
- D. Hou and T. L. Lowary, Carbohydr. Res., 2009, 344, 1911–1940 CrossRef CAS
- W. J. Morris and M. D. Shair, Org. Lett., 2008, 11, 9–12 CrossRef
- W. J. Morris and M. D. Shair, Tetrahedron Lett., 2010, 51, 4310–4312 CrossRef CAS
- R. L. Halcomb, M. D. Wittman, S. H. Olson, S. J. Danishefsky, J. Golik, H. Wong and D. Vyas, J. Am. Chem. Soc., 1991, 113, 5080–5082 CrossRef CAS
- V. G. S. Box, Heterocycles, 1982, 19, 1939–1966 CrossRef CAS
- R. R. Schmidt and J. Michel, Tetrahedron Lett., 1984, 25, 821–824 CrossRef CAS
- R. R. Schmidt, Angew. Chem., Int. Ed. Engl., 1986, 25, 212–235 CrossRef
- W. Klotz and R. R. Schmidt, Liebigs Ann. Chem., 1993, 683–690 CrossRef CAS
- S. L. Gholap, C. M. Woo, P. C. Ravikumar and S. B. Herzon, Org. Lett., 2009, 11, 4322–4325 CrossRef CAS
- Z.-X. Wang, Y. Tu, M. Frohn, J.-R. Zhang and Y. Shi, J. Am. Chem. Soc., 1997, 119, 11224–11235 CrossRef CAS
- S.-i. Hashimoto, A. Sano, H. Sakamoto, M. Hakajima, Y. Yanagiya and S. Ikegami, Synlett, 1995, 1271–1273 CrossRef CAS
- Y. Guo and G. A. Sulikowski, J. Am. Chem. Soc., 1998, 120, 1392–1397 CrossRef CAS
- R. Pongdee, B. Wu and G. A. Sulikowski, Org. Lett., 2001, 3, 3523–3525 CrossRef CAS
- G. H. Posner and S. R. Haines, Tetrahedron Lett., 1985, 26, 5–8 CrossRef CAS
- F. M. Hauser and M. Zhou, J. Org. Chem., 1996, 61, 5722–5722 CrossRef CAS
- D. Mal and N. K. Hazra, Tetrahedron Lett., 1996, 37, 2641–2642 CrossRef CAS
- V. B. Birman, Z. F. Zhao and L. Guo, Org. Lett., 2007, 9, 1223–1225 CrossRef CAS
- S. Kimura, S. Kobayashi, T. Kumamoto, A. Akagi, N. Sato and T. Ishikawa, Helv. Chim. Acta, 2011, 94, 578–591 CrossRef CAS
- W. Liu, M. Buck, N. Chen, M. Shang, N. J. Taylor, J. Asoud, X. Wu, B. B. Hasinoff and G. I. Dmitrienko, Org. Lett., 2007, 9, 2915–2918 CrossRef CAS
- P. J. Proteau, Y. Li, J. Chen, R. T. Williamson, S. J. Gould, R. S. Laufer and G. I. Dmitrienko, J. Am. Chem. Soc., 2000, 122, 8325–8326 CrossRef CAS
- S. Kim and K. H. Ahn, J. Org. Chem., 1984, 49, 1717–1724 CrossRef CAS
- S.-i. Mohri, M. Stefinovic and V. Snieckus, J. Org. Chem., 1997, 62, 7072–7073 CrossRef CAS
- K. Shin-ya, K. Furihata, Y. Teshima, Y. Hayakawa and H. Seto, Tetrahedron Lett., 1992, 33, 7025–7028 CrossRef CAS
- H. Koyama and T. Kamikawa, J. Chem. Soc., Perkin Trans. 1, 1998, 203–210 RSC
- T. E. Ballard and C. Melander, Tetrahedron Lett., 2008, 49, 3157–3161 CrossRef CAS
- C. L. Heinecke and C. Melander, Tetrahedron Lett., 2010, 51, 1455–1458 CrossRef CAS
- W. Zeng, T. E. Ballard, A. G. Tkachenko, V. A. Burns, D. L. Feldheim and C. Melander, Bioorg. Med. Chem. Lett., 2006, 16, 5148–5151 CrossRef CAS
- K. A. O'Hara, X. Wu, D. Patel, H. Liang, J. C. Yalowich, N. Chen, V. Goodfellow, O. Adedayo, G. I. Dmitrienko and B. B. Hasinoff, Free Radical Biol. Med., 2007, 43, 1132–1144 CrossRef CAS
- D. P. Arya and D. J. Jebaratnam, J. Org. Chem., 1995, 60, 3268–3269 CrossRef CAS
- R. S. Laufer and G. I. Dmitrienko, J. Am. Chem. Soc., 2002, 124, 1854–1855 CrossRef CAS
- K. S. Feldman and K. J. Eastman, J. Am. Chem. Soc., 2006, 128, 12562–12573 CrossRef CAS
- O. Khdour and E. B. Skibo, Org. Biomol. Chem., 2009, 7, 2140–2154 CAS
- H. W. Moore, Science, 1977, 197, 527–532 CAS
- C. Volkmann, E. Rössner, M. Metzler, H. Zähner and A. Zeeck, Liebigs Ann., 1995, 1995, 1169–1172 CrossRef
- J. R. Carney, S.-T. Hong and S. J. Gould, Tetrahedron Lett., 1997, 38, 3139–3142 CrossRef CAS
- S. S. Scully and J. A. Porco, Angew. Chem., Int. Ed. Engl., 2011, 50, 9722–9726 CrossRef CAS
| This journal is © The Royal Society of Chemistry 2012 |