Beyond ethylmalonyl-CoA: The functional role of crotonyl-CoA carboxylase/reductase homologs in expanding polyketide diversity
Micheal C.
Wilson
a and
Bradley S.
Moore
*ab
aScripps Institution of Oceanography, University of California at San Diego, La Jolla, California 92093-0204, USA
bSkaggs School of Pharmacy and Pharmaceutical Sciences, University of California at San Diego, La Jolla, California 92093-0204, USA. E-mail: bsmoore@ucsd.edu
First published on 29th November 2011
Abstract
Covering: up through August 2011
This review covers the emerging biosynthetic role of crotonyl-CoA carboxylase/reductase (CCR) homologs in extending the structural and functional diversity of polyketide natural products. CCRs catalyze the reductive carboxylation of α,β-unsaturated acyl-CoA substrates to produce a variety of substituted malonyl-CoA derivatives employed as polyketide synthase extender units. Here we discuss the history of CCRs in both primary and secondary metabolism, the mechanism by which they function, examples of new polyketide diversity from pathway specific CCRs, and the role of CCRs in facilitating the bioengineering novel polyketides.
 Micheal C. Wilson and Bradley S. Moore | Micheal C. Wilson earned his B.S. from the University of California at Los Angeles in 2004 with majors in Biochemistry and Marine Biology. In 2011, he was awarded his Ph.D. in Oceanography from the Scripps Institution of Oceanography for his research on the biosynthesis on ansamycin natural products from marine-derived actinomycetes under the guidance of Prof. Bradley S. Moore. He is currently studying the biosynthesis of natural products from microbial sponge symbionts as a postdoctoral research fellow in the laboratory of Prof. Jörn Piel at the University of Bonn. |
Bradley S. Moore is currently Professor of Oceanography and Pharmaceutical Sciences at the Scripps Institution of Oceanography and the Skaggs School of Pharmacy and Pharmaceutical Sciences at University of California, San Diego. He holds degrees in chemistry from the Universities of Hawaii (B.S. 1988) and Washington (Ph.D. 1994), was a postdoc at the University of Zurich (1994–1995), and held prior faculty appointments at the Universities of Washington (1996–1999) and Arizona (1999–2005). His research interests involve exploring and exploiting marine microbial genomes to discover new biosynthetic enzymes, secondary metabolic pathways, and natural products for drug discovery and development. Dr Moore is a member of the NPR Editorial Board (since 2005) and in 2011 became the NPR Editorial Board Chair. |
1 Introduction
Polyketides comprise a large class of pharmaceutically important natural products whose biological properties, including antimicrobial, immunosuppressant, and anticancer, are attributed to their specialized chemical structures derived from the condensation of simple carboxylic acid building blocks.1–3 Similar to the way in which fatty acids (FAs) are constructed, polyketide assembly progresses through successive decarboxylative Claisen condensation reactions of activated alkylmalonate precursors and acyl thioesters catalyzed by multifunctional enzymes called polyketide synthases (PKSs).4 With each addition of either a coenzyme A (CoA)-linked or acyl carrier protein (ACP)-linked PKS extender unit, the polyketide chain grows by two carbon atoms (Fig. 1). Substitutions (e.g. alkyl, hydroxy, amino) at the C2 position of the activated malonate derivative impart structural complexity to the polyketide backbone, which can organize into a seemingly infinite number of combinations.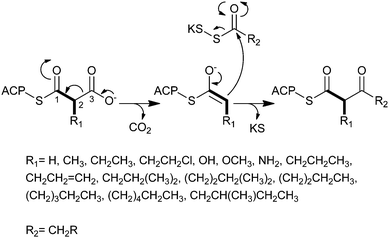 | ||
| Fig. 1 The mechanism for decarboxylative Claisen condensation in type 1 PKS elongation. | ||
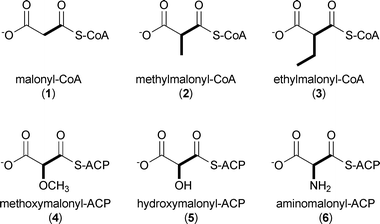
While polyketide starter units are incredibly varied in structure and size,3 extender units are much less chemically diverse.5 The most common PKS extender units include malonyl-CoA (1), methylmalonyl-CoA (2), and to a lesser extent ethylmalonyl-CoA (3) that provide H, Me, and Et substituents, respectively, to the core polyketide molecule. A small subset of PKSs additionally utilize functionalized extender units linked directly to stand-alone ACPs, in particular methoxymalonyl-ACP (4), hydroxymalonyl-ACP (5), and aminomalonyl-ACP (6), that create programmed functional group diversity in the polyketide product.5
In 2009, a new paradigm for polyketide diversification emerged involving the biosynthesis of non-traditional CoA-linked PKS extender units by the reductive carboxylation of α,β-unsaturated acyl-CoA precursors by crotonyl-CoA carboxylase/reductase (CCR) homologs.6,7 This general mechanism for the biosynthesis of atypical PKS extender units (7–17) has more than doubled the list of defined CoA-linked precursors incorporated into polyketide structures (Fig. 2). Since these substrates are CoA-tethered, CCR homologs provide a unique opportunity to genetically engineer novel “unnatural” polyketides through extender unit redesign. Herein, we review recent developments in the characterization of these enzyme biocatalysts, discuss the functional roles they play in both primary and secondary metabolism, and consider their biotechnological applications.
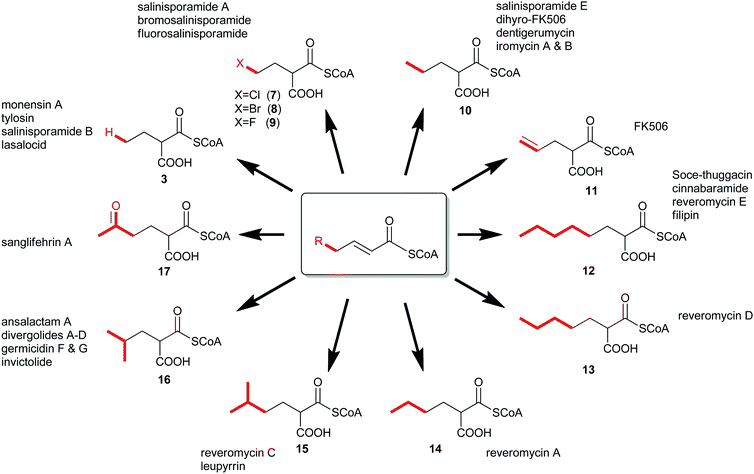 | ||
| Fig. 2 The expansion of known PKS extender units constructed by crotonyl-CoA carboxylase/reductase homologs and their associated natural products. | ||
2 Crotonyl-CoA carboxylases/reductases
CCRs and related enoyl-CoA carboxylase/reductases (ECRs) are members of the medium-chain reductase/dehydrogenase (MDR) protein superfamily that is conserved over all three domains of life.8 In addition to CCRs, the MDR superfamily includes NAD(P)-dependent alcohol dehydrogenase (ADH), quinone oxidoreductase (QOR), enoyl reductase (ER), and several other enzyme families that reduce either C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O or CC bonds. Though known to catalyze the reduction of crotonyl-CoA (18) to butyryl-CoA (19) since the 1950s,9,10 it was not until 2007 that Alber and coworkers reported that a CCR homolog from the purple non-sulfur bacterium, Rhodobacter sphaeroides, also catalyzes the reductive carboxylation of 18 to (2S)-ethylmalonyl-CoA (3).11 More intriguing was that the carboxylation was determined to be the physiologically favored reaction. This unprecedented reductive carboxylation has required a revision of the role CCR homologs in both primary and secondary metabolism.
O or CC bonds. Though known to catalyze the reduction of crotonyl-CoA (18) to butyryl-CoA (19) since the 1950s,9,10 it was not until 2007 that Alber and coworkers reported that a CCR homolog from the purple non-sulfur bacterium, Rhodobacter sphaeroides, also catalyzes the reductive carboxylation of 18 to (2S)-ethylmalonyl-CoA (3).11 More intriguing was that the carboxylation was determined to be the physiologically favored reaction. This unprecedented reductive carboxylation has required a revision of the role CCR homologs in both primary and secondary metabolism.
2.1 CCRs in primary metabolism
In the mid-1950s, while the study of FA catabolism was in full swing, FA biosynthesis remained poorly understood. Early proposals suggested that FA biosynthesis occurred by condensation of acetyl-CoA precursors by a reversal of the β-oxidation pathway, however, biochemical evidence to support this hypothesis was lacking. While studying FA biosynthesis in rat liver, Langdon reported the NADPH-dependent enzymatic reduction of crotonyl-CoA (18) to butyryl-CoA (19), the starter unit for straight chain FAs.12,13 A dialyzed soluble fraction of homogenized rat liver reduced 18 to 19 in the presence of NADPH but was not able to reduce crotonate or substitute NADH as the electron donor. Later, Langdon et al. demonstrated that hex-2-enoyl-CoA could also oxidize NADPH at rates equivalent to 18.9 Thus, Langdon termed the enzyme TPN (NADPH) ethylene reductase. Around the same time, similar experiments were carried out with pig liver by Seubert et al. in which 2-octenoyl-CoA was reduced to octanoyl-CoA in the presence of NADPH.10 In 1967, Fraser et al. purified an NADPH-dependent α,β-unsaturated ketone reductase from dog erythrocytes and human liver that could reduce 18 in addition to a variety of unsaturated ketones, including furfurylidene acetone and benzylidene actetone.14 The authors determined that the purified enzyme shared identical properties to the rat liver reductase reported by Langdon, which was also shown to reduce benzylidene actetone.14Over the next 30 years, NADPH-dependent enoyl reductase activity was reported in other mammals,15 protists,16 and bacteria17 before Reynolds and coworkers cloned and overexpressed the first bona fide CCR from Streptomyces collinus. In vitro characterization of the enzyme showed that the homodimeric S. collinus CCR catalyzed the final step in butyryl-CoA formation and was unable to accept any additional enoyl-CoA substrates or NADH as the electron donor.12 Reynolds and colleagues later showed that CCRs in other actinomycetes were critical to the flux of precursors to both the FA and polyketide biosynthetic pools.13,18–21
About a decade later, while investigating an alternate glyoxylate cycle for acetyl-CoA assimilation in R. sphaeroides, Alber and coworkers identified an NADPH-dependent enzyme that reductively carboxylates (E)-crotonyl-CoA (18) to (2S)-ethylmalonyl-CoA (3).11,22 This enzyme shared high sequence similarity (41/56% identity/similarity) to the CCR from S. collinus. In addition to the carboxylation reaction, the R. sphaeroides CCR could also reduce 18 to 19 in the absence of HCO3−/CO2, although at 1/10th the rate of the carboxylation reaction. Since Alber's initial discovery, several additional CCR homologs have been shown to also be carboxylating enzymes, thereby redefining CCRs as carboxylases with reductase activity being a remnant of their evolutionary history.23 Ethylmalonyl-CoA (3) is further processed to glycolate to enter central metabolism or can be shunted into polyketide biosynthesis by providing butyrate units, as discussed below. The genes that make up this newly defined ethylmalonyl-CoA pathway are represented in about 8% of all sequenced bacteria, only 1% of which also have a complete glyoxylate pathway as defined by Kornberg and Krebs.23–25
2.2 CCRs in secondary metabolism
The involvement of CCR in polyketide production was first reported by Reynolds and coworkers in association with the polyether antibiotic monensin A (20).26 They carried out an exhaustive study in Streptomyces cinnamonensis on the role of CCR in determining the flux of propionate and butryrate precursors into monensins A (20) and B (21).26 Disruption of the S. cinnamonensis ccr gene resulted in significant loss of 20 production while the addition of crotonic acid to wild-type cultures gave increased levels of 20. In the case of S. cinnamonensis, the encoding ccr gene is located outside the monensin biosynthetic gene cluster and thus the primary ethylmalonyl-CoA pathway must also support polyketide biosynthesis.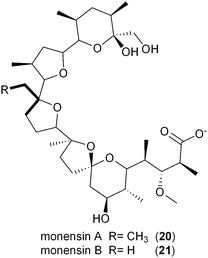
While there is more than one pathway to (2S)-ethylmalonyl-CoA (3),5 the CCR-dependent pathway appears to be the dominant source of this precursor for polyketide formation. Sequence analysis of PKS gene clusters associated with butyryl-containing polyketides supports that, unlike monensin,20 most polyketide gene clusters retain a secondary copy of the CCR encoding gene to likely synchronize 3 synthesis from acetoacetyl-CoA with polyketide assembly. These include the biosynthetic gene clusters for concanamycin (22),27 indanomycin (23),28 elaiophylin (24),29 spiramycin (25),30 tylosin (26),31 midecamycin (27),32 rosaramicin (28),33 phoslactomycin A (29),34 leptomycin B (30),35 crononatine (31),36 lasalocid A (32),37 oligomycin (33),38 tiacumicin B (34),39 tautomycetin (35),40 kirromycin (36),41 FK520 (52),42 sanglifehrin A (64), and divergolides A–D (66–69).43 Thus, although the monensin system was the first established pathway to require CCR biochemistry, it and the S. coelicolor A3(2) type III PKS germicidin synthase44 are in the minority to draw upon 3 from primary metabolism.
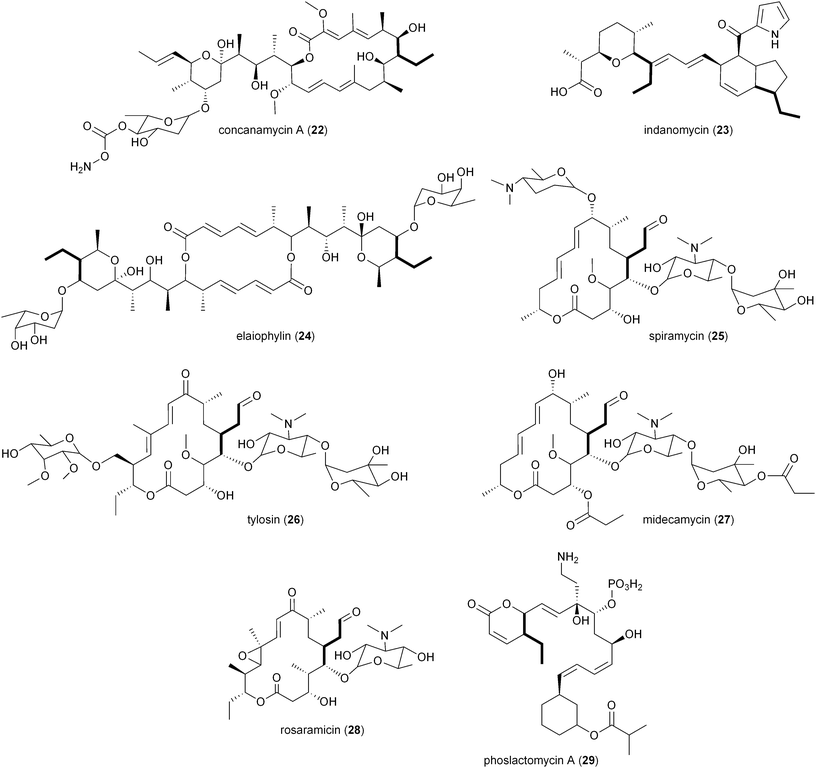
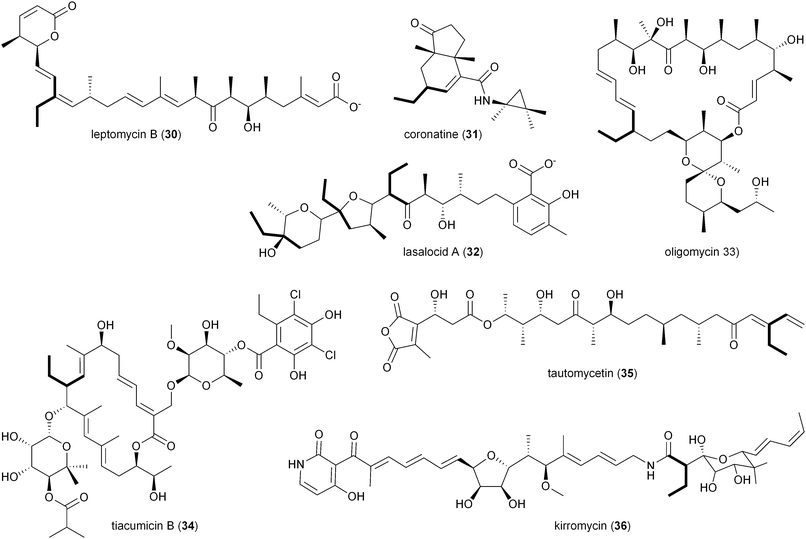
2.3 Mechanism of CCRs
The regiospecificity of the reduction reaction of CCR was first established by Robinson et al. in 1962 using tritium labeled NADPH3 incubated with 18 and enzyme preparations from rat brain and adipose tissue.45 When the isolated butyrate was degraded and analyzed, it was determined that the majority (ca. 94%) of the H3 was found on the β-carbon. The mechanism and stereochemistry of CCR's reductive carboxylation and reduction reactions were ultimately established by Alber and coworkers with recombinant R. sphaeroides CCR.46 The reductive carboxylation of (E)-crotonyl-CoA (18) to (2S)-ethylmalonyl-CoA (3) and the hydrogenation of 18 to butyryl-CoA (19) utilize NADPH as the reductant (Fig. 3a). For the carboxylation reaction it was determined that CO2 is the preferred reactive species instead of HCO3−.46 As mentioned above, reduction of 18 to 19 occurs in the absence of HCO3−/CO2, but only at a fraction of the rate of the preferred reductive carboxylation reaction. The proposed mechanism for the reductive carboxylation of α,β-unsaturated acyl-CoA substrates begins with transfer of the hydride from NADPH onto the β-carbon to give a thioester enolate, followed by electrophilic attack of the CO2 at the α-carbon. The pro-(4R) hydrogen of NADPH is transferred in both CCR reactions to the re face of the C3 position of crotonyl-CoA (Fig. 3b–c). The stereospecificity of the reduction reaction alone had previously been determined to be pro-(4S) specific for the CCR of S. collinus,47 however, a re-evaluation of the data suggested that the stereochemistry of the labeled NADPH was misassigned.46,48 Depending on whether the carboxylation or reduction reaction proceeds, either CO2 or a solvent proton are incorporated in an anti fashion at C2 of crotonyl-CoA, respectively (Fig. 3a).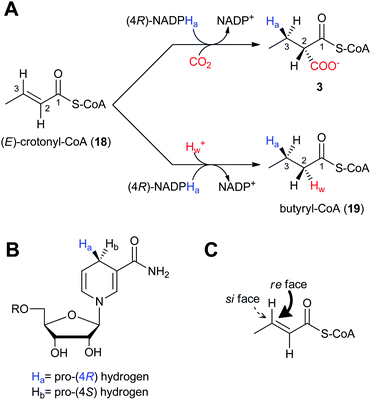 | ||
| Fig. 3 The stereochemistry and regiospecificity of CCR enzymatic reactions. A) Reductive carboxylation of (E)-crotonyl-CoA (18) to (2S)-ethylmalonyl-CoA (3) and reduction of 18 to butyryl-CoA (8). B) The pro-(4R) hydrogen (Ha) of NADPH is transferred to the C) re face of (E)-crotonyl-CoA (18). | ||
3 Polyketide diversification by CCR homologs
Sequence analysis of PKS biosynthetic gene clusters from predominantly actinomycete bacteria has recently identified a growing trend of co-clustered CCR and PKS genes involving polyketides that assimilate building blocks other than butyrate. This co-occurrence suggested that CCR homologs may use dedicated substrate derivatives in the synthesis of novel C2-modified malonyl-CoA molecules for polyketide assembly. With the first established example in 2009 of a CCR homolog involved in chloroethylmalonyl-CoA (7) biosynthesis,7 several other cases of alkylmalonyl-CoA production quickly followed over the next two years to establish CCR-based biochemistry as a major metabolic driver in PKS extender unit diversification. Phylogenetic analysis of PKS-associated CCRs shows that CCRs specific for the production of ethylmalonyl-CoA (3) largely clade separately from those responsible for production of more esoteric extender units (Fig. 4). However, unlike PKS acyltransferase (AT) or ketoacylsynthase (KS) domains, in silico prediction of the substrate specificity of CCR homologs is not readily achieved by phylogenetic analysis.49–51 This is due in part to the rare nature of some CCR substrates as well as the fact that a number of pathway specific CCR homologs can accept a variety of enoyl-CoA thioesters, as exemplified by the salinosporamide,6,52–55 FK506,56 and reveromycin57,58 series of metabolites. In this section, we highlight the recently established roles of CCR homologs in polyketide assembly.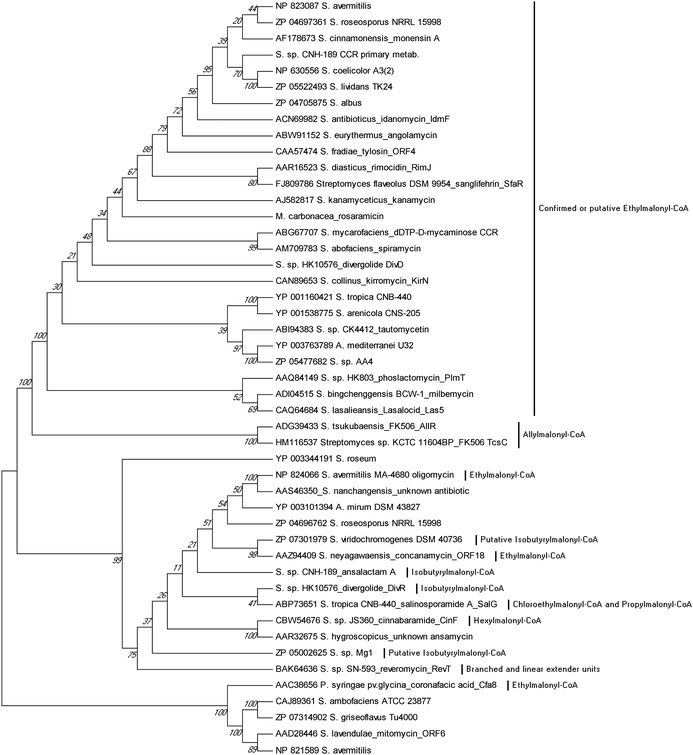 | ||
| Fig. 4 Phylogenetic analysis of CCR homologs from characterized biosynthetic gene clusters. Their evolutionary history was inferred using the UPGMA method from 1000 replicates. The analysis involved 47 protein sequences. Evolutionary analyses were conducted in MEGA5.94 | ||
3.1 Chloroethylmalonyl-CoA and bromoethylmalonyl-CoA
The salinosporamide family of potent proteasome inhibitors (37–43) from the obligate marine bacteria Salinispora tropica59 and Salinispora pacifica54 structurally differ at C2 of the γ-lactam ring, possessing H, Me, Et, Pr, EtCl, and EtBr substituents. Stable isotope tracer experiments by Moore and coworkers revealed the varied biogeneses of these biochemical precursors that were ultimately linked to a hybrid PKS–nonribosomal peptide synthetase enzymatic assembly line.52 The S. tropica salinosporamide biosynthetic gene cluster contains the CCR homolog salG that originally was hypothesized as supplying the butyrate unit for salinosporamide B assembly.7 Genetic inactivation of the salG gene, however, did not lead to significant loss in salinosporamide B production; rather, disruption of the primary metabolic CCR encoding gene strop_3612 resulted in its large reduction in yield. Deletion of salG, on the other hand, eliminated the production of the chlorinated analog salinosporamide A in which the C2 chloroethyl side chain was shown to derive from the ribose unit of S-adenosylmethionine (SAM).7,60 Further encoded within the sal biosynthetic gene cluster are seven additional genes that constitute the newly established chloroethylmalonyl-CoA pathway unique to S. tropica.7 Extensive in vivo studies utilizing gene inactivation and chemical complementation, along with in vitro biochemical and structural characterization of the gene products from the salinosporamide pathway defined the chloroethylmalonyl-CoA (7) pathway, as shown in Fig. 5a.7,61 The biosynthesis of 7 begins with the halogenation of SAM by the chlorinase SalL to yield 5′-chloro-5′-deoxyadenosine (5′-ClDA, 45). Six enzyme-catalyzed reactions convert 5′-ClDA to 4-chlorocrotonyl-CoA (46), which is subjected to reductive carboxylation by the CCR homolog SalG to yield 7. Biochemical analysis of recombinant SalG revealed its substrate preference for 4-chlorocrotonyl-CoA (46) over crotonyl-CoA (18) and its preferred reductive carboxylation turnover.7 Based on SalG's high similarity with primary CCRs, it is presumed that this class of enzymes yields the same 2S-stereochemical outcome. In addition to this natural pathway to (2S)-chloroethylmalonyl-CoA (7), SalL can also accommodate bromide ion as the nucleophilic halogen to initiate the SAM-dependent pathway to give bromoethylmalonyl-CoA (8) for PKS incorporation into bromosalinosporamide (38).6,60 The haloethylmalonyl-CoA biosynthetic pathway is unique to S. tropica and represents an orthogonal mode of polyketide halogenation that typically involves post-PKS modifying halogenating enzymes.60,62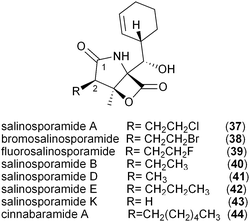
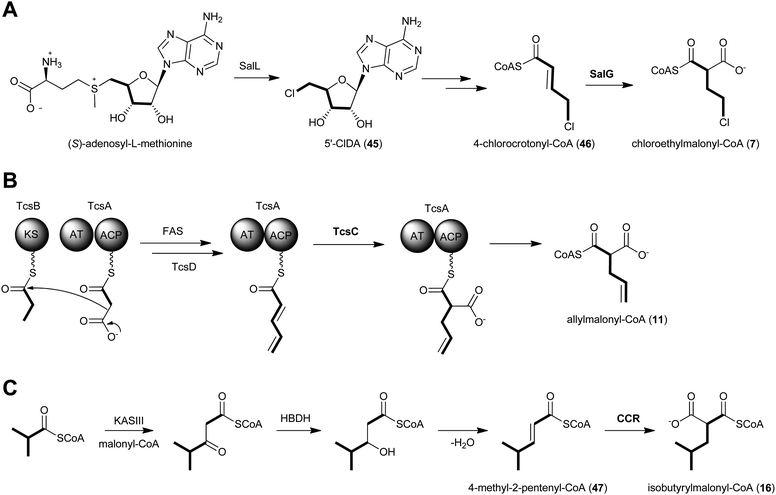 | ||
| Fig. 5 Proposed biosynthetics pathways for the dedicated PKS extender units A) chloroethylmalonyl-CoA (7), B) allylmalonyl-CoA (11) and C) isobutyrylmalonyl-CoA (16). | ||
3.2 Propylmalonyl-CoA
The genetic inactivation of the S. tropica CCR salG gene also led to a second loss in salinosporamide production, that of salinosporamide E (42), which contains a C2 propyl group.6 This observation suggested that SalG had yet another biological activity as a trans-2-pentenyl-CoA reductase/carboxylase to give (2S)-propylmalonyl-CoA (10) and that the associated SalA PKS acyltransferase (AT1) has broad substrate tolerance. Stable isotope experiments supported its biogenesis from acetate and propionate precursors, and biochemical interrogation of the SalG recombinant protein confirmed its enzymatic activity as having similar kinetics to that with 7. While SalG has broadened substrate tolerance over the co-occurring and related primary CCR Strop_3612, attempts to accommodate longer chain 2-alkenyl-CoA substrates were unsuccessful,6 yet opened the door to the possibility of related CCRs in supplying alternative PKS extender units by this strategy. While it is rare for PKSs to incorporate pentyl building blocks into their polyketide products, a few other examples include the nitric oxide synthase inhibitor iromycin A (48),63 the acyl depsipeptide dentigerumycin (49),64 and the macrolide immunosuppressant dihydro-FK506 (53).65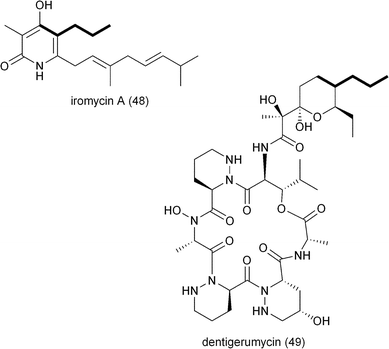
3.3 Allylmalonyl-CoA
Like the salinosporamides that owe their structural diversity and hence biological properties to PKS extender unit-derived chemistry, the FK506 family of immunosuppressant polyketides (50–53) also differs in substitution pattern at C21 of the macrolide as a result of broad PKS extender unit tolerance. In addition to the recently established allylmalonyl-CoA (11) unit that uniquely distinguishes FK506 (tacrolimus, 50),56,66 derivatives can be substituted with methylmalonyl-CoA (FK523, 51), ethylmalonyl-CoA (FK520, ascomycin, 52) or propylmalonyl-CoA (dihydro-FK506, 53).56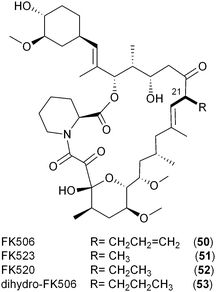
A partial biosynthetic gene cluster for FK506 was originally cloned and sequenced from Streptomyces sp. MA6548 but only contained the core PKS genes and two genes determined to be involved in post-PKS modification.65 More than a decade later, two groups independently discovered the molecular and biochemical basis for the introduction of the allyl group in FK506 from the novel PKS extender unit allylmalonyl-CoA (11).56,66 Initially reported in 2010, Petkovic and coworkers identified 15 open reading frames located upstream of the FK506 PKS core biosynthetic genes in S. tsukubaensis NRRL 18488 that were not present in the previously sequenced FK520 biosynthetic gene cluster from S. hygroscopicus var. ascomyceticus.42,66 In addition to the CCR homolog, AllR, the so-called all subcluster encodes a diketide synthase system comprised of an AT–ACP didomain protein (AllA) and an unusual ketosynthase (KS) didomain (AllK). Deletion of the KS-containing gene allK established its essential role in FK506 biosynthesis and supported a dedicated precursor pathway to 11. Further support for this pathway was achieved by chemical complementation with allylmalonyl-SNAC that restored FK506 production.66 The allylmalonyl-CoA (11) biosynthetic pathway was independently established the following year by Yoon and coworkers in a comprehensive chemical, biochemical, and genetic study of three FK506 biosynthetic gene clusters.56 In addition to identifying the four genes supporting allylmalonyl-CoA biosynthesis encoded in the tcs subcluster (synonymous to the all genes reported by Petkovic), their enzymatic functions were established through the in vitro reconstitution of the pathway (Fig. 5b). The TcsA/B (aka AllA/K) diketide synthase putatively gives rise to the β-ketopentyl-ACPTcsA product that interacts with the endogenous fatty acid synthase system to give trans-2-pentenyl-ACPTcsA. Reductive carboxylation by the CCR homolog TcsC (aka AllR) is next proposed with this ACP-bound substrate, which is a departure from previously characterized CCRs that rather function on CoA thioester-based molecules. The TcsC- and TcsD-catalyzed NADPH-dependent carboxylation and FAD-dependent dehydrogenation reactions, respectively, were monitored by ESI-MS and support the preference for ACP-based substrates in which carboxylation precedes the introduction of the double bond in the construction of allylmalonyl-ACPTcsA. The authors suggest thioester migration to allylmalonyl-CoA (11) before acyltransfer by the allylmalonyl-specific AT4 of the FK506 PKS.
While the origin of the five-carbon PKS extender unit derived from 11 and propylmalonyl-CoA (10) in the FK506 and dihydro-FK506 macrolides, respectively, originates from a dedicated diketide synthase pathway,56,66 an alternative pathway to 10 that may involve the β-oxidation of odd-chain fatty acids likely supports salinosporamide E (42) biosynthesis since the all/tcs subcluster is absent in the S. tropica genome.67 It thus remains to be seen which metabolic pathway predominates in the other pentyl-containing polyketide systems.
3.4 Hexylmalonyl-CoA and other straight chain extender units
A number of polyketides, in particular cinnabaramide A (44),55,68 Soce-thuggacin A (54),69 filipin (55),70,71 stambomycin D (59),72 and reveromycin E (63),73,74contain hexyl side chains suggestive of the PKS extender unit hexylmalonyl-CoA (12). Its biosynthesis was first proposed in 2010 by Müller and coworkers through comparative analysis of the biosynthetic gene clusters for the thuggacin macrolide antibiotics from two myxobacteria, Sorangium cellulosum So ce895 and Chondromyces crocatus Cm c5.75 The most notable difference between the products of these highly analogous pathways involves the final PKS extension in which Cmc-thuggacin A from C. crocatus incorporates a standard propionate unit derived from methylmalonyl-CoA, while Soce-thuggacin A (54) from S. cellulosum integrates an unusual octanoyl unit69 presumably derived from 12. The S. cellulosum thuggacin biosynthetic gene cluster uniquely contained the CCR homologous gene tgaD that could putatively be involved in 12 formation via the reductive carboxylation of 2-octenoyl-CoA.75 As predicted, inactivation of tgaD yielded a 54-negative mutant supporting the central role of TgaD in its construction. Unlike the previous examples involving the salinosporamide and FK506 systems in which the primary metabolites methyl- and ethylmalonyl-CoA could chemically complement the pathway specific CCR mutant to give natural product analogs, no such recovery was observed in the thuggacin system suggesting that the gatekeeper AT11 domain is specific toward the longer chain hexylmalonate, which is consistent with the absence of natural C2 variants in S. cellulosum. However, due to TgaD's relatively low similarity to characterized CCR homologs, a more complete biochemical characterization is needed to confirm its anticipated function and to determine whether it operates with CoA- or ACP-linked substrates.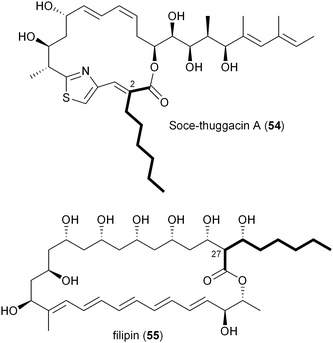
A second example of hexylmalonyl-CoA (12) as a PKS extender unit involves cinnabaramide A (44) from the terrestrial Streptomyces species strain JS360 that belongs to the salinosporamide family of proteosome inhibitors.68 The cinnabaramides are distinguished from the salinoporamides by the putative incorporation of 12 by AT2 of the cinnabaramide PKS, CinA. As proposed in Soce-thuggacin A (54) biosynthesis, 12 is similarly derived from the reductive carboxylation of 2-octenoyl-CoA by a pathway specific CCR, which in this case is encoded by cinF. Interestingly, the sequence similarity between CinF and TgaG is very low considering that they are predicted to catalyze the same reaction and may rather reflect their different taxonomic origins. As with the thuggacins, gene inactivation of cinF generated a mutant incapable of producing the cinnabaramides, prompting Müller and coworkers to conclude that CinF is responsible for 12 production in Streptomyces sp. JS360.55
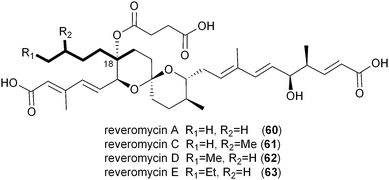
While the Soce-thuggacins and cinnabaramides strictly maintain hexyl side chains in their polyketide structures, the reveromycin family (60–63) of osteoclast-targeting polyketides contains four natural C18 variants suggestive of a mixture of alkylmalonyl-CoA precursors (12–15).73,76,77 Though the current biosynthetic literature by Osada and coworkers is focused on reveromycin's novel spiroketal formation,58 an equally fascinating feature involves the incorporation of four rare or unique PKS extender units in a single position on their structures. The reveromycins putatively incorporate butyryl- (14), pentyl- (13), isopentyl- (15), and hexylmalonyl-CoA (12) precursors as the fourth extender unit of their polyketide backbone. As expected based on the pathways described thus far, the reveromycin biosynthetic gene cluster contains a CCR homolog (revT) with potentially promiscuous substrate specificity.
The first in vitro characterization of a 2-octenoyl-CoA carboxylase/reductase was reported in 2011 by Kwon and coworkers with the CCR homolog PteB from Streptomyces avermitilis.70 The pteB gene is located within the biosynthetic gene cluster of filipin (55),38 an antifungal polyene macrolide originally isolated in 1955 that incorporates an octanoate unit during the final PKS extension.70,71 Recombinant PteB had dual activity, reducing 2-octenoyl-CoA to octanoyl-CoA in the absence of bicarbonate, and in its presence, reductively carboxylating it to 12. PteB exhibited broad substrate tolerance toward crotonyl-, 2-hexenoyl-, 2-octenoyl-, and 2-decenoyl-CoAs, consistent with structure analogs, such as chainin,78 strevertene,79 and takanawaene,80 from other actinomycetes that differ in the nature of the final extender unit. Though gene clusters for the 55 analogs have not been reported, their conserved structures suggest a common biosynthetic pathway with modified substrate specificity of a CCR and associated gatekeeper AT domain to account for differing side-chains at C27.
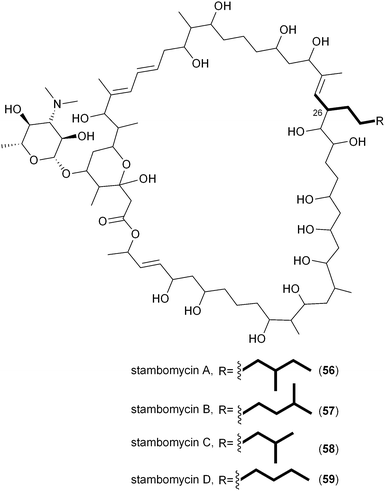
In a departure from the CCR-based biosynthesis of 12, Streptomyces ambofaciens appears to generate its dedicated alkylmalonyl-CoA substrates for stambomycin assembly by an alternate route.72 DNA sequence analysis of the 150 kb stambomycin biosynthetic gene cluster failed to identify a pathway specific CCR homolog; instead Laureti et al. observed the gene samR0483 that encodes a biotin-dependent carboxylase related to propionyl-CoA carboxylase that may function to directly carboxylate saturated octanoyl-CoA and derivatives to ultimately provide stambomycin D (59) and other C26 structure analogs.
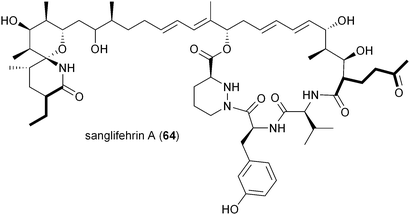
The biosynthesis of the potent cyclophilin inhibitor sanglifehrin A (64) was recently established through the sequence analysis of its biosynthetic machinery in Streptomyces flaveolus to reveal an intriguing possibility of the oxidized PKS extender unit 2-oxobutylmalonyl-CoA (17).81 Liu and colleagues suggest its biosynthesis from the dedicated iterative type I PKS SfaK to make a putative unsaturated triacetate intermediate for reductive carboxylation to 2-oxobutylmalonyl-CoA; however, the authors have not identified a specific CCR for this reaction as found in the allylmalonyl-CoA (11) pathway that also involves a separate PKS for its construction. While the sanglifehrin biosynthesis gene cluster does indeed harbor a CCR gene, sfaR, it rather functions as a bone fide CCR responsible for 3 biosynthesis linked to the formation of the proposed PKS primer (2R)-ethylmalonamyl-CoA.
3.5 Isobutyrylmalonyl-CoA and other branched chain extender units
A series of polyketides independently isolated and characterized in early 2011 by the Moore and Hertweck groups suggested the involvement of the novel branched extender unit isobutyrylmalonyl-CoA (16) and its synthesis via a CCR mechanism. The discovery of the ansa macrolide natural product ansalactam A (65) from marine-derived Streptomyces sp. CNH-189 suggested an unusual branched side chain at C22 that was proposed to originate from the reductive carboxylation of 4-methyl-2-pentenyl-CoA (47).82 Moore and coworkers explored its biogenesis with stable isotopes and ruled out a leucine origin of this moiety that rather is derived from the condensation of isobutyrate and acetate to 4-methyl-2-pentenoate.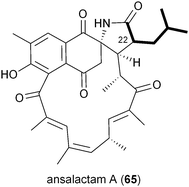
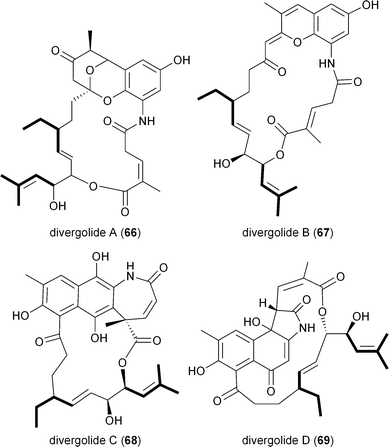
Hertweck and coworkers similarly isolated a novel group of ansa macrolide antibiotics, divergolides A–D (66–69), from the mangrove endophyte Streptomyces sp. HKI0576 that also contain the unsaturated version of the isobutyryl polyketide side chain.83 The polyketide backbone that contains this branched substituent is disrupted with an oxygen atom that could originate via a Baeyer–Villiger-type oxidation reaction. Precursor experiments with D7-isobutyrate not only labeled 66–69 as anticipated but also identified two new germicidin derivatives, F (70) and G (71), originating from this branched building block.43 Shotgun sequence analysis provided the divergolide biosynthetic gene cluster and illuminated the molecular basis for isobutyrylmalonyl-CoA (16) biosynthesis (Fig. 5c). Two copies of CCR-encoding genes were identified in the type I PKS gene cluster, one (DivD) suggested for ethylmalonyl-CoA (3) biosynthesis and the second (DivR) for isobutyrylmalonyl-CoA (16) assembly. The divR gene resides in a three-gene cassette that also includes the FabH-like KS divS and the 3-hydroxybutyryl-CoA dehydrogenase divT that together encodes 16 synthesis (Fig. 5c) as supported by its heterologous expression with the germicidin type III PKS in Streptomyces albus to give 70 and 71.
Recently two new analogs related to the queen recognition pheromone invictolide were described by Sato and coworkers from a marine-derived actinomycete most closely related to Nocardiopsis tangguensis.84 The tetrapropionate δ-lactone 3,6,7-tri-epi-invictolide differs from the analog 72 in the fourth and final ketide residue that hypothetically derives instead from 16.
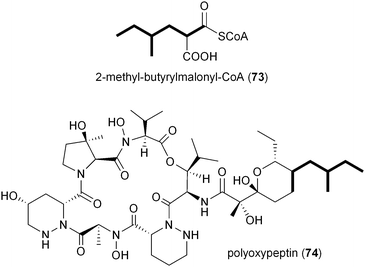
Inspection of publicly available genomic information suggests that the isobutyrylmalonyl-CoA (16) gene cassette resides in a number of orphan biosynthetic systems, suggesting a greater frequency of this newly described PKS extender unit in actinomycetes (Fig. 6). Related branched-chain extender units, such as the proposed 2-methyl-butyrylmalonyl-CoA (73) and isopentylmalonyl-CoA (15), may be synthesized in a similar FabH-based fashion to give polyoxypeptin A (74)85 and reveromycin C (61),58 respectively. In the case of 61, a FabH-like KS, revR, and CoA-ligase, revS, are located directly upstream of the reveromycin CCR, revT. Similar to 65–72, this three gene cassette is proposed to function in the biosynthesis of the unusual extender units of the reveromycins (12–15).58
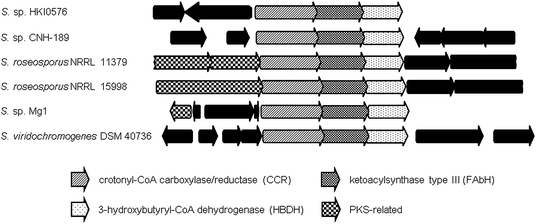 | ||
| Fig. 6 Orthologous gene neighborhoods to the isobutyrylmalonyl-CoA (16) pathway of the divergolide (66–69) producer, Streptomyces sp. HKI0576, putatively involved in the biosynthesis of branched PKS extender units. | ||
4 Engineering new polyketides utilizing pathway specific CCR homologs
In the majority of cases involving CCR-based PKS extender units, the associated acyltransferase (AT) domain displays relaxed specificity toward its selection of malonate substrates. This observation is evident from the number of derivatives of the salinosporamide (37–43), FK506 (50–53), monensin (20–21), reveromycin (60–63), germicidin (71–72), and thuggacin (54) natural product series. Previous phylogenetic analyses of AT domains suggest that ATs with the same substrate specificity share a common evolutionary origin with malonyl- and methylmalonyl-specific ATs clustering into distinct clades (Fig. 7).86,87 The less common ethylmalonyl-specific AT, however, does not form a distinct clade but rather resides in subclades within the methylmalonyl group, which is suggestive that this group may have evolved more than once. Examination of the haloethylmalonyl-preferred SalA AT17 and the allylmalonyl-preferred FkbB AT488 associated with the CCR homologs SalG and TcsC, respectively, similarly placed them in the methylmalonyl clade. A more thorough phylogenetic analysis of the AT domains associated with the CCR-derived PKS substrates highlighted in this review article is shown in Fig. 7. Similar to AT domains that accept ethylmalonyl-CoA (3), our analysis shows that AT domains specific for less common CCR-derived extender units also reside in subclades within the methylmalonyl group. Moreover, AT domains specific for the same atypical extender unit may have also evolved separately. This is perhaps best exemplified by the AT domains specific for hexylmalonyl-CoA in the cinnabaramide, thuggacin, and filipin pathways (Fig. 7).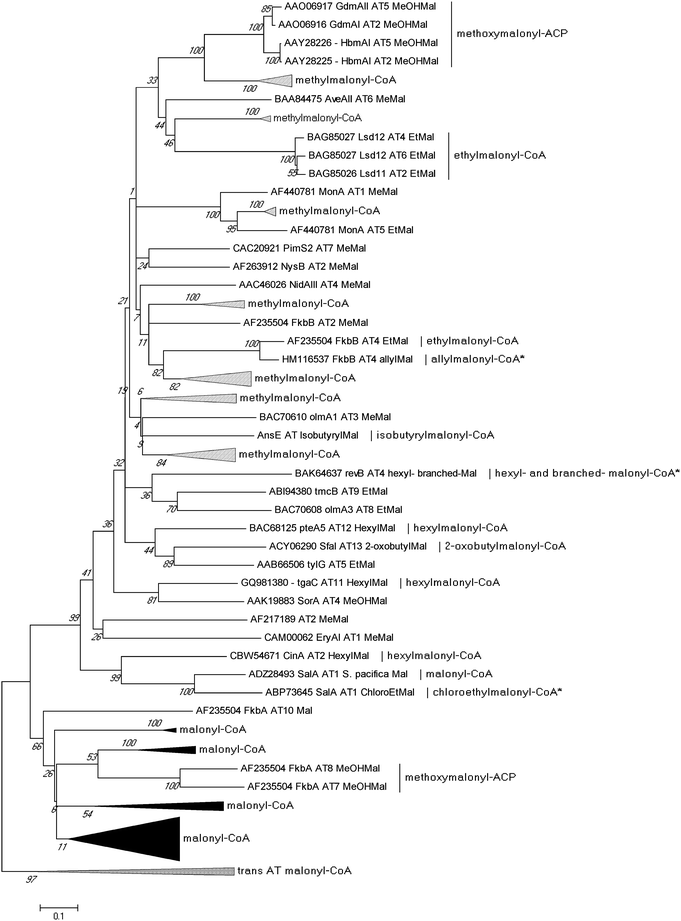 | ||
| Fig. 7 Phylogenetic analysis of 130 acyltransferase (AT) domains from characterized and putative PKS proteins. The evolutionary history was inferred using the Neighbor-Joining method, and the bootstrap consensus tree was generated from 1000 replicates. Evolutionary analyses were conducted in MEGA5.94 Asterisks (*) indicate that AT domains accept multiple extender units. | ||
Given the apparent relaxed natural specificity of ATs associated with CCR-derived substrates, several investigators exploited this enzymatic promiscuity to selectively bioengineer novel structure analogs via mutasynthesis and genetic engineering methods (Fig. 8). Moore and coworkers first expanded salinosporamide biosynthesis to give the unnatural derivative fluorosalinosporamide (39), a very potent “reversible” proteasome inhibitor, through mutasynthesis with the salL chlorinase knockout mutant supplemented with 5′-fluoro-5′-deoxyadenosine.89 By complementing the mutant with simple 4-substituted crotonates, they later could rescue the natural biosynthesis of salinosporamides A (37), B (40), E (42), and bromosalinosporamide (38) as well as create the unnatural 39.6 Fluorosalinosporamide (39) was also genetically engineered by replacing the salL chlorinase gene with the flA fluorinase gene from Streptomyces cattleya, representing the first engineered biosynthetic experiment to generate a fluoroorganic molecule from inorganic fluoride.90
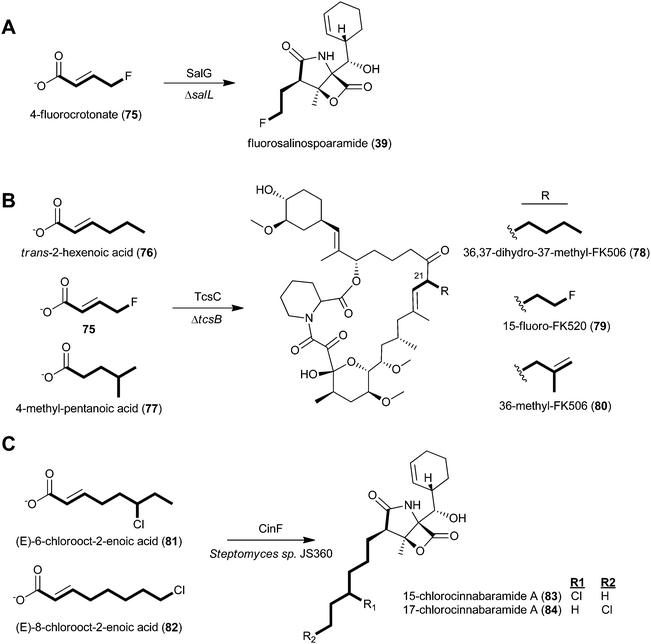 | ||
| Fig. 8 Engineering of natural product analogs through chemical complementation of A) the S. tropica chlorinase (salL) deficient mutant to generate fluorosalinosporamide (39), B) the Streptomyces sp. KCTC 11604BP tcsB mutant to generate three unnatural FK506 analogs (78–80), and C) the wild-type cinnabaramide producer, Streptomyces sp. JS630, to generate chlorinated cinnabaramide analogs (83–84). | ||
Relaxed substrate specificity was also observed in the FK506 and cinnabaramide series. The genetic disruption of the tcsB KS gene prevented de novo allylmalonyl-CoA biosynthesis and allowed for the mutasynthesis of FK506 analogs derived from a series of carboxylic acids.56 Specifically, trans-2-hexenoic acid (76), 4-fluorocrotonic acid (75), and 4-methylpentanoic acid (77) provided 36,37-dihydro-37-methyl-FK506 (78), 15-fluoro-FK520 (79), and 36-methyl-FK506 (80), respectively. The latter molecule, 80, is noteworthy in two regards: first, the unnatural precursor was presumably modified by the TcsD dehydrogenase to introduce the double bond, as in FK506 (50), and second, 80 showed improved neurite outgrowth activity. With the discovery of fluoroethylmalonyl-CoA (9)90 and isobutyrylmalonyl-CoA (16)83 biosynthetic pathways, the genetic engineering of 79 and 80, respectively, is now feasible. In the case of cinnabaramide diversification, Müller and coworkers took a precursor-directed biosynthesis approach with the wild-type strain since the background synthesis of the proposed natural substrate 2-octenoyl-CoA furnished from fatty acid metabolism could not be eliminated.55 Administration of (E)-6-chlorooct-2-enoic acid (81), (E)-8-chlorooct-2-enoic acid (22), and their corresponding N-acetylcysteamine thioesters resulted in new cinnabaramide derivatives with 15- and 17-chloro groups (83–84) that on average exhibited superior bioactivity over the parent cinnabaramide.
5 Outlook
The 2007 functional revision of R. sphaeroides CCR as a reductive carboxylase11 followed by the 2009 discovery of the S. tropica CCR homolog SalG as having alternate substrate specificity6,7 exposed a new strategy in PKS extender unit diversification from α,β-unsaturated acyl thioesters. Several CCR-mediated pathways to linear and branched-chain (halo)alkylmalonyl-CoA molecules have since been uncovered, making these related pathways, although rare, the most functionally accommodating in providing diverse PKS substrates5 (Fig. 2). To date, all characterized pathways originate from actinomycetes91 and myxobacteria,92 arguably two of the most metabolically rich groups of prokaryotes. Additional pathways are undoubtedly awaiting discovery not just from these bacterial groups, but also from others based on genomic scanning of publicly available sequences. For instance, CCRs are widespread amongst the proteobacteria and are also present in the genomes of firmicutes and spirochaetes. Even more intriguing is that the genome of the firmicute Ruminococcus sp. 18P13 contains a CCR (CBL16354) that is located directly upstream of a FabH-like ketoacylsynthase and adjacent to genes predicted to be involved in polyketide and non-ribosomal peptide assembly. To the best of our knowledge, no secondary metabolite has been associated with these biosynthetic genes.The polyketide engineering potential has only been scratched with the successful analoging in the salinosporamide,6 cinnabaramide,55 and FK50656 series in which the broad natural substrate tolerance of native CCRs and associated gatekeeper ATs accommodated modest changes in precursor structure. Whether more ambitious modifications in substrate or in product design are biochemically feasible with rationally engineered biosynthetic systems is to be seen. Early AT engineering in the all-methylmalonate erythromycin PKS allowed for the successful swap of an ethylmalonate unit,93 thereby illuminating the concept of extender unit remodeling. To address the renewed engineering potential with alkylmalonyl-CoAs will require a firmer understanding of the structural and mechanistic basis of substrate selectivity and binding in CCR homologs and in alkylmalonyl-specific ATs. Based on the variety of substrates accommodated by CCR and AT homologs, structure-based enzyme engineering is anticipated to provide active site changes that correlate to substrate selectivity. Its application toward designer polyketide molecules is significant since the longer acyl chains in CCR-derived PKS extender units provide greater structural design flexibility as seen in the reactive chemical handles of the haloethylmalonyl and allylmalonyl series that engender key biological features to their polyketide products. Precursor supply of the α,β-unsaturated acyl thioester substrates will also have to be addressed as the pathways so far elucidated show that these molecules come from diverse metabolic origins involving dedicated, as well as general, pathways. Hence the outlook for the further diversification of the ethylmalonyl-CoA pathway in polyketide biosynthesis and bioengineering is promising with new insight likely right around the corner.
6 Acknowledgements
We appreciate generosity of the NIH through grant CA127622 to support our exploration of PKS extender unit chemistry in salinosporamide biosynthesis.7 References
- J. Staunton and K. J. Weissman, Nat. Prod. Rep., 2001, 18, 380–416 RSC.
- B. Shen, Curr. Opin. Chem. Biol., 2003, 7, 285–295 CrossRef CAS.
- B. S. Moore and C. Hertweck, Nat. Prod. Rep., 2002, 19, 70–99 RSC.
- M. A. Fischbach and C. T. Walsh, Chem. Rev., 2006, 106, 3468–3496 CrossRef CAS.
- Y. A. Chan, A. M. Podevels, B. M. Kevany and M. G. Thomas, Nat. Prod. Rep., 2009, 26, 90–114 RSC.
- Y. Liu, C. Hazzard, A. S. Eustáquio, K. A. Reynolds and B. S. Moore, J. Am. Chem. Soc., 2009, 131, 10376–10377 CrossRef CAS.
- A. S. Eustáquio, R. P. McGlinchey, Y. Liu, C. Hazzard, L. L. Beer, G. Florova, M. M. Alhamadsheh, A. Lechner, A. J. Kale, Y. Kobayashi, K. A. Reynolds and B. S. Moore, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 12295–12300 CrossRef.
- H. Riveros-Rosas, A. Julian-Sanchez, R. Villalobos-Molina, J. P. Pardo and E. Pina, Eur. J. Biochem., 2003, 270, 3309–3334 CrossRef CAS.
- R. G. Langdon, J. Biol. Chem., 1957, 226, 615–629 CAS.
- W. Seubert, G. Greull and F. Lynen, Angew. Chem., 1957, 69, 359–361 CrossRef CAS.
- T. J. Erb, I. A. Berg, V. Brecht, M. Mueller, G. Fuchs and B. E. Alber, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 10631–10636 CrossRef CAS.
- R. G. Langdon, J. Am. Chem. Soc., 1955, 77, 5190–5192 CrossRef CAS.
- K. K. Wallace, B. Zhao, H. A. McArthur and K. A. Reynolds, FEMS Microbiol. Lett., 1995, 131, 227–234 CrossRef CAS.
- I. M. Fraser, M. A. Peters and M. G. Hardinge, Mol. Pharmacol., 1967, 3, 233–247 CAS.
- S. K. Maitra and S. Kumar, J. Biol. Chem., 1974, 249, 111–117 CAS.
- H. Inui, K. Miyatake, Y. Nakano and S. Kitaoka, J. Biochem., 1986, 100, 995–1000 CAS.
- K. K. Wallace, Z.-Y. Bao, H. Dai, R. Digate, G. Schuler, M. K. Speedie and K. A. Reynolds, Eur. J. Biochem., 1995, 233, 954–962 CAS.
- K. Akopiants, G. Florova, C. Li and K. A. Reynolds, J. Ind. Microbiol. Biotechnol., 2006, 33, 141–150 CrossRef CAS.
- L. Han and K. A. Reynolds, J. Bacteriol., 1997, 179, 5157–5164 CAS.
- H. Liu and K. A. Reynolds, J. Bacteriol., 1999, 181, 6806–6813 CAS.
- H. Liu and K. A. Reynolds, Metab. Eng., 2001, 3, 40–48 CrossRef CAS.
- B. E. Alber, R. Spanheimer, C. Ebenau-Jehle and G. Fuchs, Mol. Microbiol., 2006, 61, 297–309 CrossRef CAS.
- B. E. Alber, Appl. Microbiol. Biotechnol., 2011, 89, 17–25 CrossRef CAS.
- T. J. Erb, G. Fuchs and B. E. Alber, Mol. Microbiol., 2009, 73, 992–1008 CrossRef CAS.
- H. L. Kornberg and H. A. Krebs, Nature, 1957, 179, 988–991 CrossRef CAS.
- D. O'Hagan, S. V. Rogers, G. R. Duffin and K. A. Reynolds, J. Antibiot., 1995, 48, 1280–1287 CAS.
- S. F. Haydock, A. N. Appleyard, T. Mironenko, J. Lester, N. Scott and P. F. Leadlay, Microbiology, 2005, 151, 3161–3169 CrossRef CAS.
- C. Li, K. E. Roege and W. L. Kelly, ChemBioChem, 2009, 10, 1064–1072 CrossRef CAS.
- S. F. Haydock, T. Mironenko, H. I. Ghoorahoo and P. F. Leadlay, J. Biotechnol., 2004, 113, 55–68 CrossRef CAS.
- F. Karray, E. Darbon, N. Oestreicher, H. Dominguez, K. Tuphile, J. Gagnat, M.-H. Blondelet-Rouault, C. Gerbaud and J.-L. Pernodet, Microbiology, 2007, 153, 4111–4122 CrossRef CAS.
- A. R. Gandecha, S. L. Large and E. Cundliffe, Gene, 1997, 184, 197–203 CrossRef CAS.
- N. Midoh, S. Hoshiko and T. Murakami, 2006, United States of America patent no. 7795001.
- C. M. Farnet, A. Staffa and X. Yang, US patent, 10/205,032, 2003.
- N. Palaniappan, B. S. Kim, Y. Sekiyama, H. Osada and K. A. Reynolds, J. Biol. Chem., 2003, 278, 35552–35557 CrossRef CAS.
- Z. Hu, R. Reid and H. Gramajo, J. Antibiot., 2005, 58, 625–633 CrossRef CAS.
- D. Bignell, R. Seipke, J. Huguet-Tapia, A. Chambers, R. Parry and R. Loria, Mol. Plant-Microbe Interact., 2010, 23, 161–175 CrossRef CAS.
- A. Migita, M. Watanabe, Y. Hirose, K. Watanabe, T. Tokiwano, H. Kinashi and H. Oikawa, Biosci., Biotechnol., Biochem., 2009, 73, 169–176 CrossRef CAS.
- S. Omura, H. Ikeda, J. Ishikawa, A. Hanamoto, C. Takahashi, M. Shinose, Y. Takahashi, H. Horikawa, H. Nakazawa, T. Osonoe, H. Kikuchi, T. Shiba, Y. Sakaki and M. Hattori, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 12215–12220 CrossRef CAS.
- Y. Xiao, S. Li, S. Niu, L. Ma, G. Zhang, H. Zhang, J. Ju and C. Zhang, J. Am. Chem. Soc., 2011, 133, 1092–1105 CrossRef CAS.
- W. Li, Y. Luo, J. Ju, S. Rajski, H. Osada and B. Shen, J. Nat. Prod., 2009, 72, 450–459 CrossRef CAS.
- T. Weber, K. J. Laiple, E. K. Pross, A. Textor, S. Grond, K. Welzel, S. Pelzer, A. Vente and W. Wohlleben, Chem. Biol., 2008, 15, 175–188 CrossRef CAS.
- K. Wu, L. Chung, W. P. Revill, L. Katz and C. D. Reeves, Gene, 2000, 251, 81–90 CrossRef CAS.
- Z. Xu, L. Ding and C. Hertweck, Angew. Chem., Int. Ed., 2011, 50, 4667–4670 CAS.
- L. Song, F. Barona-Gomez, C. Corre, L. Xiang, D. W. Udwary, M. B. Austin, J. P. Noel, B. S. Moore and G. L. Challis, J. Am. Chem. Soc., 2006, 128, 14754–14755 CrossRef CAS.
- J. D. Robinson, R. O. Brady and C. R. Maxwell, J. Lipid Res., 1962, 3, 243–245 CAS.
- T. J. Erb, V. Brecht, G. Fuchs, M. Muller and B. E. Alber, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 8871–8876 CrossRef CAS.
- H. Liu, K. K. Wallace and K. A. Reynolds, J. Am. Chem. Soc., 1997, 119, 2973–2979 CrossRef CAS.
- S. B. Mostad, H. L. Helming, C. Groom and A. Glasfeld, Biochem. Biophys. Res. Commun., 1997, 233, 681–686 CrossRef CAS.
- G. Yadav, R. S. Gokhale and D. Mohanty, J. Mol. Biol., 2003, 328, 335–363 CrossRef CAS.
- G. Yadav, R. S. Gokhale and D. Mohanty, Nucleic Acids Res., 2003, 31, 3654–3658 CrossRef CAS.
- G. Yadav, R. S. Gokhale and D. Mohanty, PLoS Comput. Biol., 2009, 5, e1000351 Search PubMed.
- L. L. Beer and B. S. Moore, Org. Lett., 2007, 9, 845–848 CrossRef CAS.
- K. A. Reed, R. R. Manam, S. S. Mitchell, J. Xu, S. Teisan, T. H. Chao, G. Deyanat-Yazdi, S. T. Neuteboom, K. S. Lam and B. C. Potts, J. Nat. Prod., 2007, 70, 269–276 CrossRef CAS.
- A. S. Eustáquio, S.-J. Nam, K. Penn, A. Lechner, M. C. Wilson, W. Fenical, P. R. Jensen and B. S. Moore, ChemBioChem, 2011, 12, 61–64 CrossRef.
- S. Rachid, L. Huo, J. Herrmann, M. Stadler, B. Kopcke, J. Bitzer and R. Muller, ChemBioChem, 2011, 12, 922–931 CrossRef CAS.
- S. Mo, D. H. Kim, J. H. Lee, J. W. Park, D. B. Basnet, Y. H. Ban, Y. J. Yoo, S. W. Chen, S. R. Park, E. A. Choi, E. Kim, Y. Y. Jin, S. K. Lee, J. Y. Park, Y. Liu, M. O. Lee, K. S. Lee, S. J. Kim, D. Kim, B. C. Park, S. G. Lee, H. J. Kwon, J. W. Suh, B. S. Moore, S. K. Lim and Y. J. Yoon, J. Am. Chem. Soc., 2011, 133, 976–985 CrossRef CAS.
- L. Fremlin, M. Farrugia, A. M. Piggott, Z. Khalil, E. Lacey and R. J. Capon, Org. Biomol. Chem., 2011, 9, 1201–1211 CAS.
- S. Takahashi, A. Toyoda, Y. Sekiyama, H. Takagi, T. Nogawa, M. Uramoto, R. Suzuki, H. Koshino, T. Kumano, S. Panthee, T. Dairi, J. Ishikawa, H. Ikeda, Y. Sakaki and H. Osada, Nat. Chem. Biol., 2011, 7, 461–468 CrossRef CAS.
- T. A. M. Gulder and B. S. Moore, Angew. Chem., Int. Ed., 2010, 49, 9346–9367 CrossRef CAS.
- A. S. Eustáquio, F. Pojer, J. P. Noel and B. S. Moore, Nat. Chem. Biol., 2008, 4, 69–74 CrossRef.
- A. J. Kale, R. P. McGlinchey and B. S. Moore, J. Biol. Chem., 2010, 285, 33710–33717 CrossRef CAS.
- F. H. Vaillancourt, E. Yeh, D. A. Vosburg, S. Garneau-Tsodikova and C. T. Walsh, Chem. Rev., 2006, 106, 3364–3378 CrossRef CAS.
- F. Surup, O. Wagner, J. von Frieling, M. Schleicher, S. Oess, P. Müller and S. Grond, J. Org. Chem., 2007, 72, 5085–5090 CrossRef CAS.
- D.-C. Oh, M. Poulsen, C. R. Currie and J. Clardy, Nat. Chem. Biol., 2009, 5, 391–393 CrossRef CAS.
- H. Motamedi and A. Shafiee, Eur. J. Biochem., 1998, 256, 528–534 CAS.
- D. Goranovic, G. Kosec, P. Mrak, S. Fujs, J. Horvat, E. Kuscer, G. Kopitar and H. Petkovic, J. Biol. Chem., 2010, 285, 14292–14300 CrossRef CAS.
- D. W. Udwary, L. Zeigler, R. N. Asolkar, V. Singan, A. Lapidus, W. Fenical, P. R. Jensen and B. S. Moore, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 10376–10381 CrossRef CAS.
- M. Stadler, J. Bitzer, A. Mayer-Bartschmid, H. Muller, J. Benet-Buchholz, F. Gantner, H. V. Tichy, P. Reinemer and K. B. Bacon, J. Nat. Prod., 2007, 70, 246–252 CrossRef CAS.
- H. Steinmetz, H. Irschik, B. Kunze, H. Reichenbach, G. Höfle and R. Jansen, Chem.–Eur. J., 2007, 13, 5822–5832 CrossRef CAS.
- H.-G. Yoo, S.-Y. Kwon, S. Kim, S. Karki, Z.-Y. Park and H.-J. Kwon, Biosci., Biotechnol., Biochem., 2011, 75, 1191–1193 CrossRef CAS.
- B. Berkoz and C. Djerassi, Proc. Chem. Soc., London, 1959, 316–317 CAS.
- L. Laureti, L. Song, S. Huang, C. Corre, P. Leblond, G. L. Challis and B. Aigle, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 6258–6263 CrossRef CAS.
- L. Fremlin, M. Farrugia, A. M. Piggott, Z. Khalil, E. Lacey and R. J. Capon, Org. Biomol. Chem., 2011, 9, 1201–1211 CAS.
- S. Takahashi, A. Toyoda, Y. Sekiyama, H. Takagi, T. Nogawa, M. Uramoto, R. Suzuki, H. Koshino, T. Kumano, S. Panthee, T. Dairi, J. Ishikawa, H. Ikeda, Y. Sakaki and H. Osada, Nat. Chem. Biol., 2011, 7, 461–468 CrossRef CAS.
- K. Buntin, H. Irschik, K. J. Weissman, E. Luxenburger, H. Blocker and R. Muller, Chem. Biol., 2010, 17, 342–356 CrossRef CAS.
- H. Koshino, H. Takahashi, H. Osada and K. Isono, J. Antibiot., 1992, 45, 1420–1427 CAS.
- J. T. Woo, M. Kawatani, M. Kato, T. Shinki, T. Yonezawa, N. Kanoh, H. Nakagawa, M. Takami, K. H. Lee, P. H. Stern, K. Nagai and H. Osada, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 4729–4734 CrossRef CAS.
- R. C. Pandey, N. Narasimhachari, K. L. RinehartJr. and D. S. Millington, J. Am. Chem. Soc., 1972, 94, 4306–4310 CrossRef CAS.
- J. Guo, G. Schlingmann, G. T. Carter, K. Nakanishi and N. Berova, Chirality, 2000, 12, 43–51 CrossRef CAS.
- T. Fukuda, Y. P. Kim, K. Iizima, H. Tomoda and S. Omura, J. Antibiot., 2003, 56, 454–458 CAS.
- X. Qu, N. Jiang, F. Xu, L. Shao, G. Tang, B. Wilkinson and W. Liu, Mol. BioSyst., 2011, 7, 852–861 RSC.
- M. C. Wilson, S. J. Nam, T. A. Gulder, C. A. Kauffman, P. R. Jensen, W. Fenical and B. S. Moore, J. Am. Chem. Soc., 2011, 133, 1971–1977 CrossRef CAS.
- L. Ding, A. Maier, H. H. Fiebig, H. Gorls, W. H. Lin, G. Peschel and C. Hertweck, Angew. Chem., Int. Ed., 2011, 50, 1630–1634 CrossRef CAS.
- S. Sato, F. Iwata, S. Yamada and H. Kawahara, J. Antibiot., 2011, 64, 385–389 CrossRef CAS.
- K. Umezawa, Y. Ikeda, O. Kawase, H. Naganawa and S. Kondo, J. Chem. Soc., Perkin Trans. 1, 2001, 1, 1550–1553 RSC.
- H. Jenke-Kodama, A. Sandmann, R. Muller and E. Dittmann, Mol. Biol. Evol., 2005, 22, 207–2039 CrossRef.
- C. P. Ridley and C. Khosla, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 4595–4600 CrossRef CAS.
- H. Motamedi and A. Shafiee, Eur. J. Biochem., 1998, 256, 528–534 CAS.
- A. S. Eustáquio and B. S. Moore, Angew. Chem., Int. Ed., 2008, 47, 3936–3938 CrossRef.
- A. S. Eustáquio, D. O'Hagan and B. S. Moore, J. Nat. Prod., 2010, 73, 378–382 CrossRef.
- M. Nett, H. Ikeda and B. S. Moore, Nat. Prod. Rep., 2009, 26, 1362–1384 RSC.
- S. C. Wenzel and R. Müller, Nat. Prod. Rep., 2009, 26, 1385–1407 RSC.
- D. L. Stassi, S. J. Kakavas, K. A. Reynolds, G. Gunawardana, S. Swanson, D. Zeidner, M. Jackson, H. Liu, A. Buko and L. Katz, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 7305–7309 CrossRef CAS.
- K. Tamura, D. Peterson, N. Peterson, G. Stecher, M. Nei and S. Kumar, Mol. Biol. Evol., 2011, 28, 2731–2739 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2012 |